
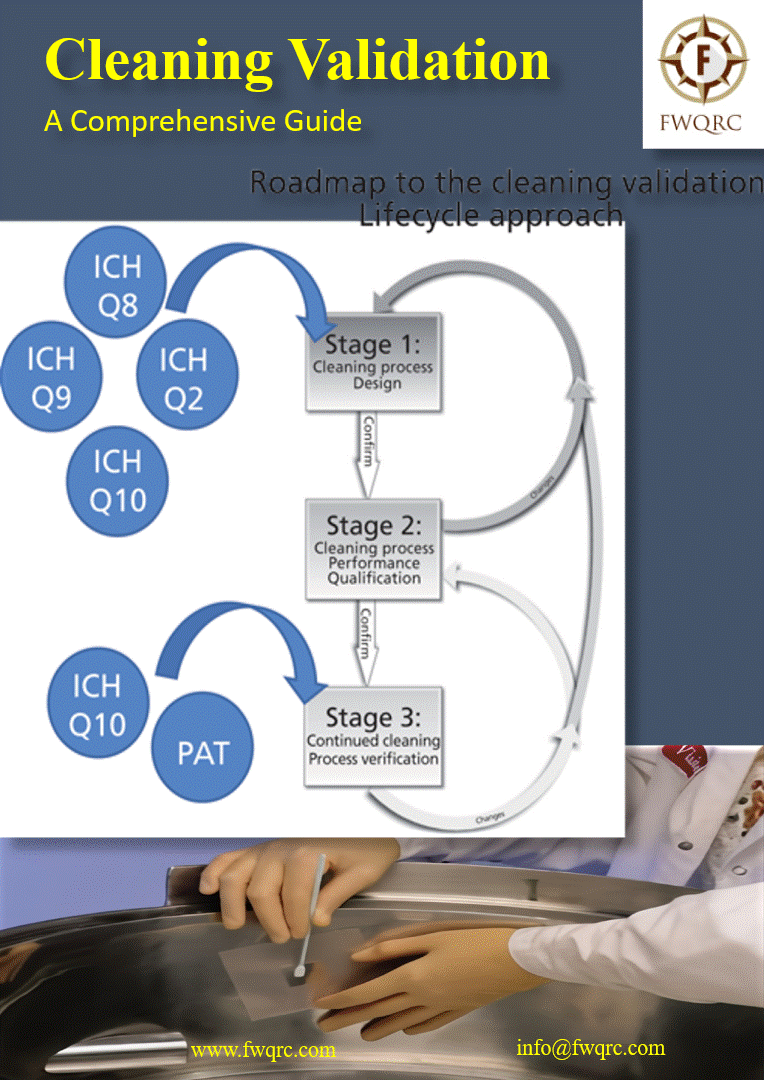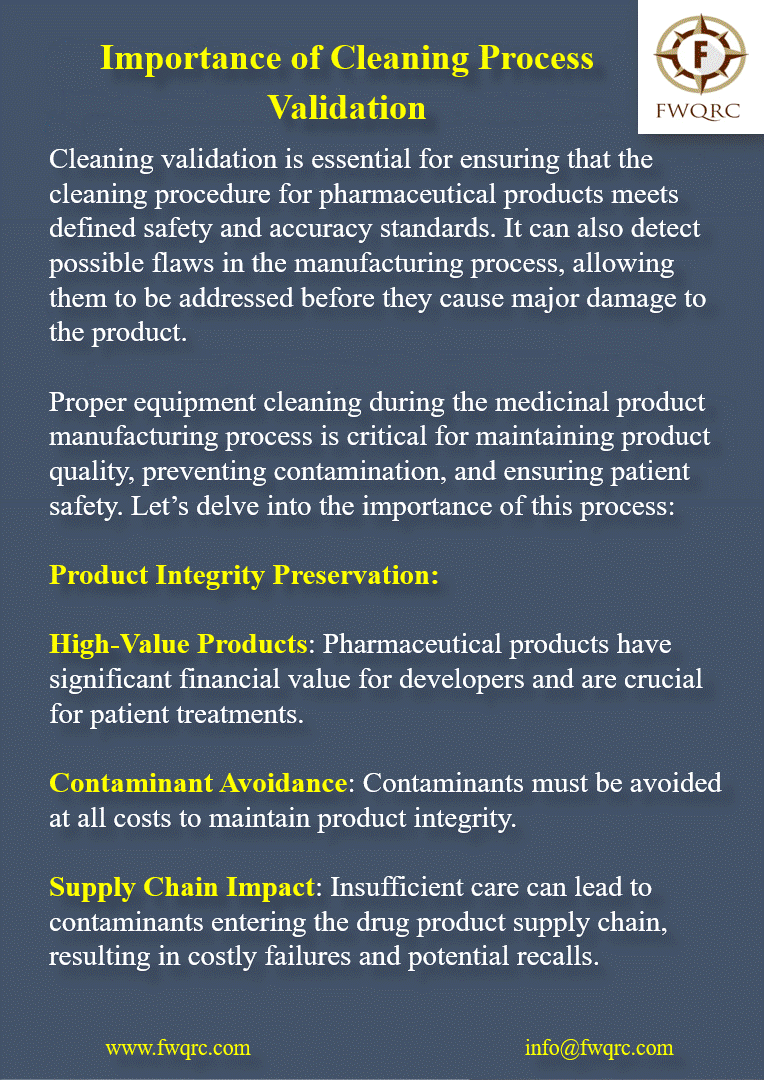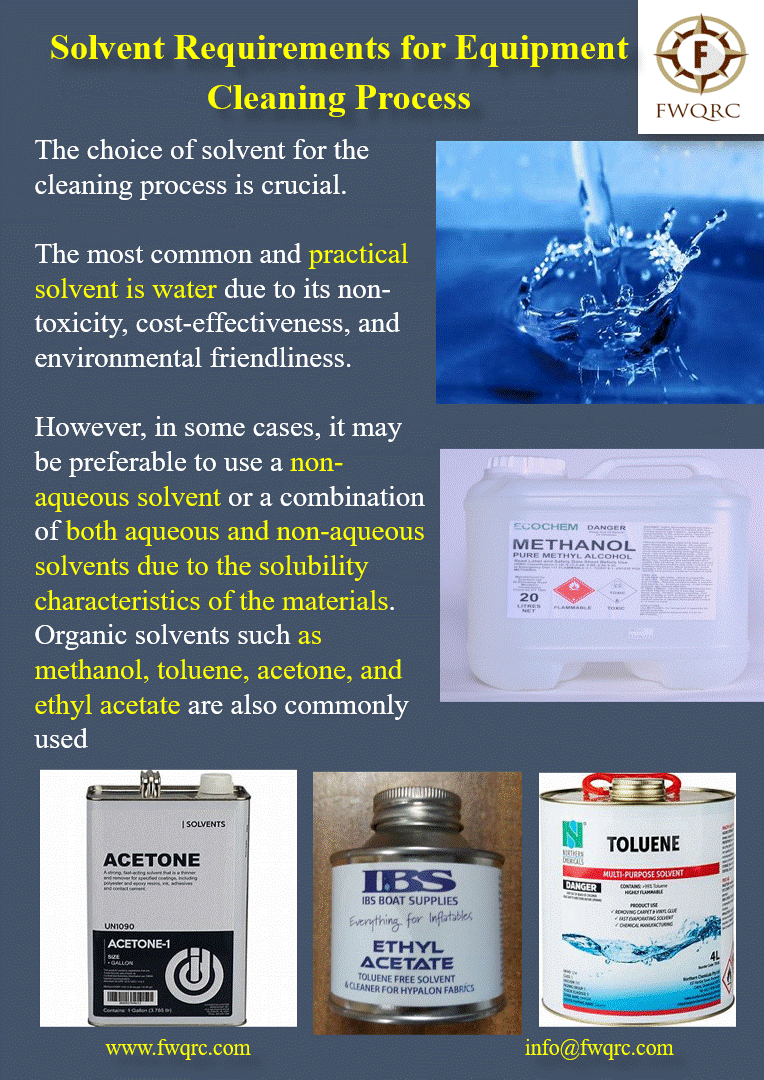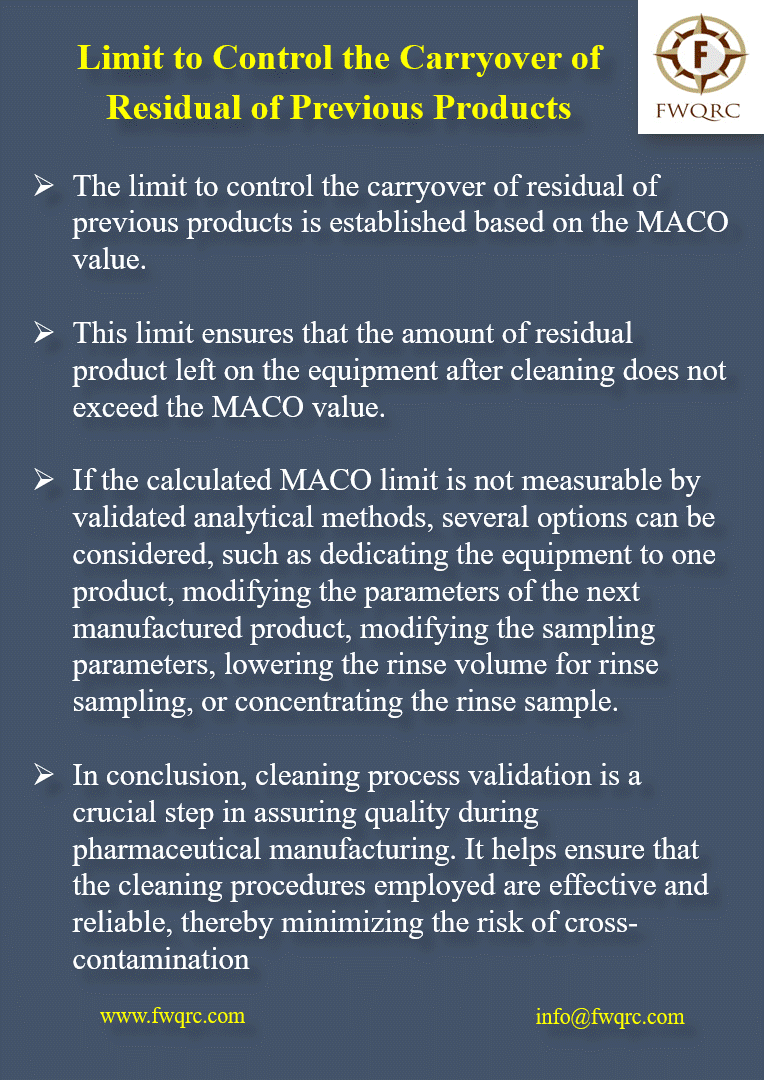
Cleaning process validation is a critical aspect of pharmaceutical manufacturing. It ensures that the cleaning procedures employed between different production runs are effective and reliable, minimizing the risk of cross-contamination.
What is Cleaning Process Validation?
Cleaning process validation is a scientific technique that provides documented, risk-based evidence that a typical cleaning method will consistently clean the equipment or a medical device in compliance with its predetermined specifications and quality attributes.
It ensures that previous product traces have been removed, operations are contamination-free, and safe batch-to-batch transitions are possible.
Importance of Cleaning Process Validation:
Cleaning validation is essential for ensuring that the cleaning procedure for pharmaceutical products meets defined safety and accuracy standards. It can also detect possible flaws in the manufacturing process, allowing them to be addressed before they cause major damage to the product.
Proper equipment cleaning during the medicinal product manufacturing process is critical for maintaining product quality, preventing contamination, and ensuring patient safety. Let’s delve into the importance of this process:
Product Integrity Preservation:
High-Value Products: Pharmaceutical products have significant financial value for developers and are crucial for patient treatments.
Contaminant Avoidance: Contaminants must be avoided at all costs to maintain product integrity.
Supply Chain Impact: Insufficient care can lead to contaminants entering the drug product supply chain, resulting in costly failures and potential recalls.
Cross-Contamination Prevention:
Mono-Product Processes: For processes involving a single product, visual inspection alone is often adequate for cleaning validation or verification.
Multi-Product Processes: In multi-product operations, the risk of cross-contamination is higher.
Scientific Approach: Cleaning limits based on a scientific approach (such as Health Based Exposure Limits) must be set.
Shared Equipment Train: These limits should be integrated into the shared equipment train.
Control Alignment: Control levels (including sampling and detection methods) should align with the product type and equipment visibility.
Equipment Cleaning Practices:
Changeover Cleaning:
Equipment used for different products during campaigns should undergo thorough cleaning.
Interval Cleaning:
Regular cleaning during a campaign, as necessary.
Dedicated End-of-Campaign Cleaning:
Ensures equipment is ready for the next batch.
Remember, effective equipment cleaning on direct or indirect product contact surfaces is essential to prevent cross-contamination and maintain product quality throughout the manufacturing process.
Solvent Requirements for Equipment Cleaning Process:
The choice of solvent for the cleaning process is crucial.
The most common and practical solvent is water due to its non-toxicity, cost-effectiveness, and environmental friendliness.
However, in some cases, it may be preferable to use a non-aqueous solvent or a combination of both aqueous and non-aqueous solvents due to the solubility characteristics of the materials. Organic solvents such as methanol, toluene, acetone, and ethyl acetate are also commonly used.
Analytical Test Method Requirements:
Analytical methods used in the cleaning validation process should be validated according to the requirements of regulatory bodies like the FDA and EMA.
The four most common types of analytical methods, each with its own set of validation requirements, are identity tests, quantitative tests for impurity content, limit tests for the control of impurities, and potency tests.
The detection limit for each analytical method should be sufficiently sensitive to detect the established acceptable level of the residue or contaminant.
Procedure for Establishing MACO Values:
Maximum Allowable Carryover (MACO) refers to the acceptable amount of a product that can be carried over from one batch to the next.
The correct calculation of MACO is vital for the integrity and success of the cleaning validation program.
MACO can be calculated using various industry-recognized approaches such as health-based, therapeutic, toxicological, and 10 parts per million (ppm) methods.
For example, one common formula for calculating MACO using Permitted Daily Exposure (PDE) is:
MACO = {PDE x MBS}/{MDD}
where:
MACO: Maximum allowable carryover in mg
PDE: Permitted daily exposure of the previous product
MBS: Minimum batch size for the next product in mg
MDD: Maximum Daily dose of the next product in mg
Limit to Control the Carryover of Residual of Previous Products:
The limit to control the carryover of residual of previous products is established based on the MACO value.
This limit ensures that the amount of residual product left on the equipment after cleaning does not exceed the MACO value.
If the calculated MACO limit is not measurable by validated analytical methods, several options can be considered, such as dedicating the equipment to one product, modifying the parameters of the next manufactured product, modifying the sampling parameters, lowering the rinse volume for rinse sampling, or concentrating the rinse sample.
In conclusion, cleaning process validation is a crucial step in assuring quality during pharmaceutical manufacturing. It helps ensure that the cleaning procedures employed are effective and reliable, thereby minimizing the risk of cross-contamination.
#food #drugs #cosmetics #medicaldevices #medicinalproducts #api #qa #qc #ra #reugulatoryaffairs #qualitycompliance #regulatorycompliance #trendingnow #trendingposts #riskanakysis #gapanalysis #riskassessment #capa #remediation #auditor #leadauditor #irca #cqi #pharamceuticals #industries #clv #amv #cleaningvalidation #analyticalmethodvalidation #csv