News Quality Management: A Triad Approach by FWQRC-Global GxP & Regulatory Consultant Read More » May 15, 2024 No Comments
Blog Final Rule to Collect Antimicrobial Sales and Distribution Information by Animal Species Read More » May 11, 2024 No Comments
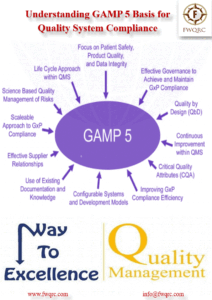
Blog Unravelling the Concepts of GAMP 5 for Effective Computer System Validation Read More » May 5, 2024 No Comments
Blog Understanding Medicinal Product Mock Recalls & related Regulations Read More » May 3, 2024 No Comments
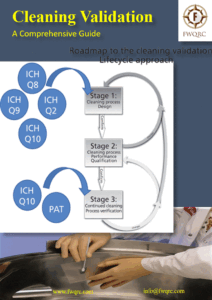
Blog Cleaning Process Validation in Medicinal Product Manufacturing Read More » April 28, 2024 No Comments
Blog Modernization of Cosmetics Regulation Act of 2022 (MoCRA)-A comprehensive guide Read More » April 26, 2024 No Comments