

All the posts related to Life Sciences and its requirements shall be updated by the technical team on daily basis. Everyone is open to review and share your comments regarding the posts.

QUALITY PROFESSIONALS
Hi, Greetings from FWQRC……………..
Today’s topic is about how Quality professionals help organisations to deliver. We explain who they are and how they go about it
- Everyone in an organisation is responsible for quality – from the CEO to the intern. But not everyone can be a quality expert. It’s important to have people who can provide the knowledge, tools and guidance to help everyone else play their part in pursuing excellence.These people are called quality professionals. Their job is to make sure organisations deliver.
- Quality professionals come in many guises. Some are generalists, some are specialists. Many will have titles such as quality manager, quality engineer, quality director or assurance manager, while others deal with quality as part of a broader remit. Some are concerned with the delivery of products and services, while some are part of the leadership of an organisation. Some are employed in-house, while others work outside the organisations they deal with.
- What unites quality professionals is their dedication to protecting and strengthening their organisations by making sure that stakeholders’ needs are met – and ideally, that their expectations are exceeded.

What quality professionals do
To put quality at the heart of their organisations, quality professionals focus on three specific areas, or competencies:
- Strong governance: This starts with top management expressing a commitment to quality. Effective governance means making sure that the aims of management are crystal clear, that they reflect the requirements of stakeholders, and that the right people, policies and processes are in place to turn them into action.
- Proper assurance: This ensures that the policies and priorities that have been decided on are being carried out properly, and that whatever is being produced – whether it’s a product, service, or project – is meeting stakeholders’ needs.
- A culture of improvement: This means continually evaluating the organisation’s performance to improve efficiency, eliminate waste, reduce risk, respond to changes and create new opportunities.
The measure of a quality professional’s success is how well we
- Protect reputation: avoiding the potentially catastrophic risks of getting things wrong
- Enhance reputation: maximizing value for our customers and stakeholders
- Improve profitability: eliminating unnecessary cost and waste and growing revenue
- Drive change: contributing to the ongoing improvement of the organisation
Quality professionals are recognized by colleagues as
- Agents for change: transforming processes, behaviour and culture
- Guardians: protecting the business by identifying appropriate standards for business performance and assuring that they are met
- Collaborators: working closely with leaders and managers
- Leaders: creating, managing and improving the organisation’s business process systems
- Progressive: understanding the realities of managing organisations in dynamic environments
- Holistic: looking across business functions and hierarchies to advocate a broad process and customer-centric view of the organisation
- Professional at FWQRC: qualified by professional institute (CQI), the CQI, and bound by a rigorous code of conduct.
Thank you for viewing FWQRC blogs….

MITIGATING RISKS WITH BLACK CHAIN TECHNOLOGY
Hi, Greetings from FWQRC……….
This blog is related to how block chain contributes to drug supply management

WHAT IS BLOCK CHAIN?
Block chain, or a distributed ledger, is a way of organisation information is a way that gives all appropriate parties access to the information they need and keeps that information secure from people who should not see it
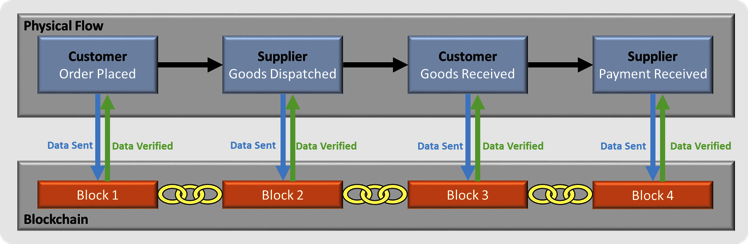
In terms of quality, block chains can ensure that every part of a supply chain can have assurance that the materials & products moving through it have reached a particular standard passed checks and compiled with necessary regulations
In its simple form,block chain is information that is shared across a group of computers so that if one person updates that information others are able to see it
HOW BLOCK CHAIN CONTRIBUTES TO DRUG SUPPLY-CHAIN MANAGEMENT
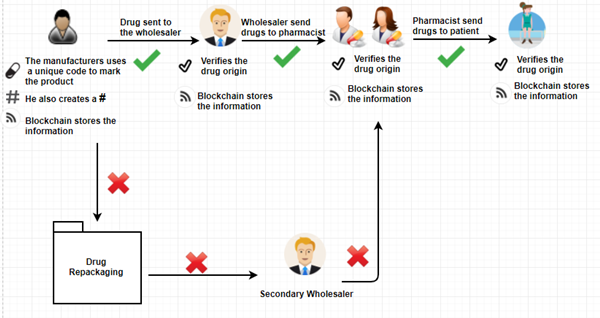
Imagine a simple supply chain: Company A produces raw material;company B makes it into a product,while company C sells it. With block chain,company A can alert company B & company C of changes in supply chain-such as overproduction-that they can then use to moderate their manufacturing process. Company B could extend their working hours and sales strategy, while company C could plan a marketing campaign to move the extra product
Sharing decentralized information in this way means that business relationships will become much more flexible,benefiting the participants and requiring no outside help. This can be a highly effective means of self regulation
HOW BUSINESSES ARE USING BLOCK CHAIN IN THEIR SUPPLY CHAIN:
Global research firm, Gartner, predicts that by 2023,some 30% of manaufacturing complanies with revenue of more than $5bn will be using block chain to drive down costs and improve tracebility and transperancy
Block chain as a strategy will force companies to look beyond the boundaries of their own firm & establish shared process and consensus mechanisms with their supply chain partners
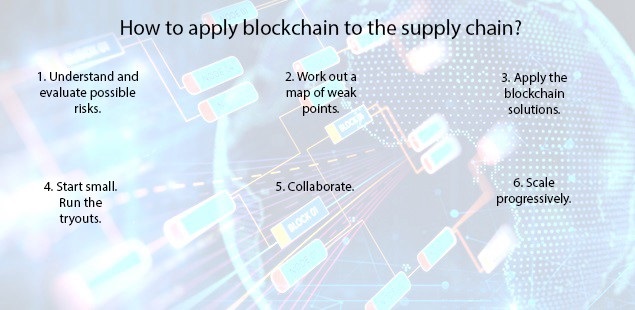
CHALLENGES OF THE MODEL
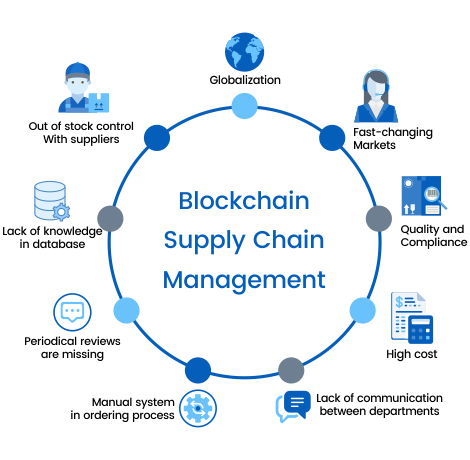
- The benefits are great,but they may come at a cost
- Block chain represents a challenge for businesses “Companies that have an ageing information technology infrastructure will struggle to interact effectively with digitally native companies
- Tech companies have a responsibility to make the user experience as easy & seamless as possible for everyone in the supply chain

GMP, GUIDELINES, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
Small Entity Compliance Guide (SECG)
February 3, 2020
The Food and Drug Administration today announced the availability of a Small Entity Compliance Guide (SECG) to help packaged food manufacturers meet federal standards in the final rule “ Food Labeling: Revision of the Nutrition and Supplement Facts Labels.” The final rule, which was published on May 27, 2016, amends the labeling regulations for foods and dietary supplements to provide updated nutrition information on the label to help consumers maintain healthy dietary practices.
The SECG is aimed at small businesses and restates, in plain language and in a question and answer format, the provisions in the final rule. It includes the following sections in addition to references:
Who is subject to the rule?
What foods are covered by the rule?
What foods are not covered by the rule?
Which nutrients must newly be declared, and what changes have been made to nutrients previously required or allowed to be declared?
How do I comply with recordkeeping requirements?
How have the values of nutrients been updated?
How do I comply with the formatting requirements?’
When must I comply with the rule?
Why must I comply with the rule?
Compliance with the updated Nutrition Facts labeling regulations was required by January 1, 2020, for manufacturers with $10 million or more in annual food sales, while manufacturers with less than $10 million in annual food sales will have an additional year to comply. During the first 6 months following the January 1, 2020, compliance date, FDA plans to work cooperatively with manufacturers to meet the new Nutrition Facts label requirements and will not focus on enforcement actions regarding these requirements during that time. FDA intends to exercise enforcement discretion to give manufacturers of single-ingredient sugars such as honey and maple syrup, and certain cranberry products, until July 1, 2021, to comply.
For Compliance,please write to fwqrcservices@gmail.com

QUALITY CULTURE, REGULATORY FOCUS NEWS LETTER, YOUR PARTNER
DEVELOPMENT OF A QUALITY CULTURE
Hi, Greetings from FWQRC……
Today’s topic is about the development of a Quality Culture and the supporting role of international standard development

Quality Culture is a term being discussed with increasing regulatory among quality professionals as well as more widely outside of the quality profession, as organisations and governments consider continuous challenges including productivity, competitive strategy and reputation protection.Within a quality culture, everyone is involved in the ongoing pursuit of excellence

Pharmaceutical culture of quality
Each worker or team is both a supplier & customer within their organisation and takes responsibility for the quality of their output. In this setting, there is no need to check the final products as an incremental review has occurred on an ongoing basis at each stage
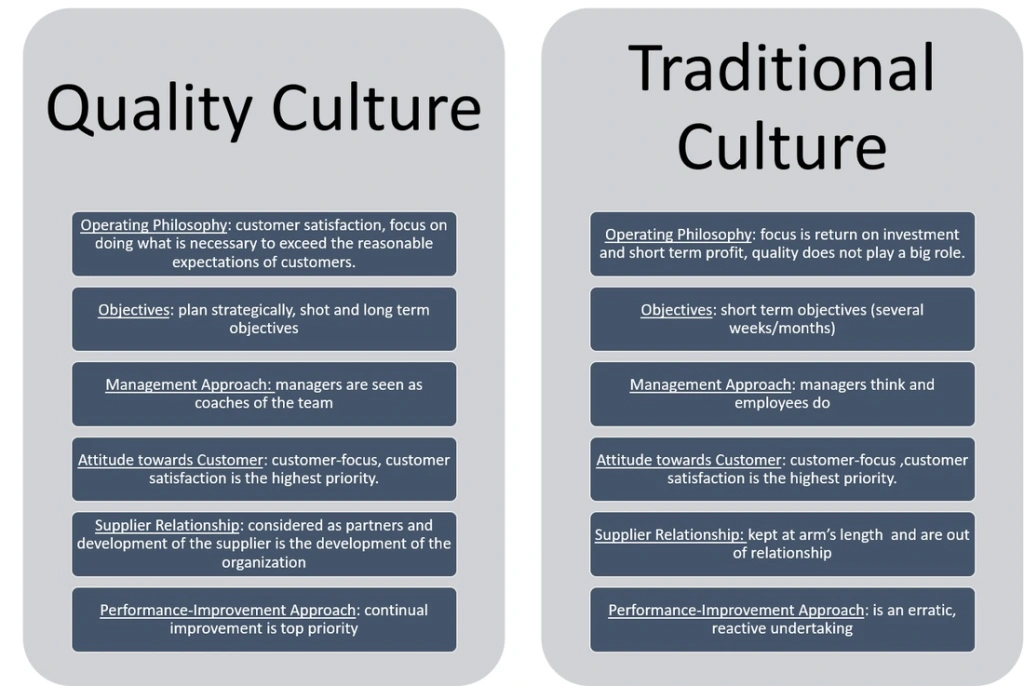
Conversely, the absence of a quality culture is likely to be characterized by an absence of individual or team responsibility and the need for costly & time consuming inspection and test regimes which add little value
Conclusion: Given the more elusive nature of culture compared to other facts of organization life, it is not unreasonable for individuals to look for external sources of guidance. Assistance may be required to understand current culture, determine that culture which might best address requirements, and to help define & deliver actions to support the quality culture journey.
Conclusion: Given the more elusive nature of culture compared to other facts of organisation life, it is not unreasonable for individuals to look for external sources of guidance. Assistance may be required to understand current culture, determine that culture which might best address requirements and to help define & deliver actions to support the quality culture journey.

Write to fwqrcservices@gmail.com to understand your organisations current culture, determine that culture and deliver actions to support the quality culture

AGRICULTURE, FOOD, REGULATORY FOCUS NEWS LETTER, YOUR PARTNER
FDA, USDA and EPA announce joint platform to streamline information about agricultural biotechnology products
Hi,Welcome to FWQRC Regulatory Focus News Letter…

Today, in recognition of January 2020 as National Biotechnology Month, the U.S. Food and Drug Administration, the Department of Agriculture and the Environmental Protection Agency launched a Unified Website for Biotechnology Regulation. The website streamlines information about the three regulatory agencies charged with overseeing agriculture biotechnology products and is part of President Donald J. Trump’s Executive Order on Modernizing the Regulatory Framework for Agricultural Biotechnology Products.
“This is a time of unprecedented scientific innovation. Agricultural biotechnology promises to bring dynamic new products to the marketplace,” said FDA Commissioner Stephen Hahn, M.D. “At the FDA, we are committed to fostering flexible, risk-based approaches in this field while upholding our mission of protecting and promoting both human and animal health and animal well-being, for example by reducing their susceptibility to diseases like novel influenzas and resistance to zoonotic or foreign animal diseases. Our approach balances our internationally respected, science-based review standards with our ongoing risk-based regulatory approaches to ensure the safety of our food supply.”
The Unified Website for Biotechnology Regulation describes the federal review process for certain biotechnology products and allows users to submit questions to the three agencies. The goals of this website are to provide enhanced customer service to innovators and developers, while ensuring Americans continue to enjoy the safest and most affordable food supply in the world and can learn more about the safe use of biotechnology innovations.
In October 2018, the FDA announced its Plant and Animal Biotechnology Innovation Action Plan, which focuses on the agency’s risk-based regulatory approach. This approach will help secure confidence in the reliability and performance of plant and animal-based innovative products for consumers and America’s global trading partners. Making sure these products are safe is critical to maintaining consumer and commercial confidence in them and will help them to realize their full potential benefits for human and animal health.

The FDA uses a flexible, risk-based approach to the oversight of plant- and animal- derived products of biotechnology, focusing on safety and, where applicable, effectiveness. The agency’s approach includes, when appropriate, updating and clarifying science-based policies to support innovation and ensure that our regulatory processes are efficient, predictable, and proportionate to risk.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.

COSMETICS, GMP, HEALTHCARE INSTITUTIONS, LIFE SCIENCES, MEDICAL DEVICES, REGULATORY FOCUS NEWS LETTER
Cosmetic Registration Reports

Hi, Welcome to FWQRC Regulatory Focus News Letter
About Blog FWQRC Regulatory focus pharma news, views and analysis of healthcare in a rapidly changing world. Not only do we keep you connected with the latest trends in pharma, we can also help you develop and bring to life your own thoughts, ideas and inspirations to enable you and your business to become key pharma influencers
The information in the tables below is a monthly report of activity in FDA’s Voluntary Cosmetic Registration Program (VCRP)
VCRP Monthly Status Report
Because the law does not require cosmetic firms to register their establishments or file their product formulations with FDA, participation in this program is voluntary. For this reason, the information below does not indicate the total number of companies manufacturing or marketing cosmetics in this country, or all cosmetic products on the market.
Activity for December 2019
- Number of online accounts activated this month: 103
- Number of products filed this month: 593
- Total activity since the launch of the new online system, September 20, 2018
- Number of active online accounts: 2,816
- Number of active cosmetic establishment registrations: 1,395
- Number of labelers that have filed product formulations*: 650
- Number of filed products: 8,333
- Number of product formulations discontinued**: 373
- Number of product formulations rejected***: 958
- Total activity since the VCRP was established, in 1972
- Number of active cosmetic establishment registrations: 4,392
- Number of labelers that have filed product formulations*: 3,071
- Number of active products on file: 68,838
- Number of product formulations discontinued**: 82,094
- Number of product formulations rejected***: 5,101


GMP, HEALTHCARE INSTITUTIONS, LIFE SCIENCES, MEDICAL DEVICES, PHARMA
FWQRC™ intended to provide a decision pathway for drug & medical devices manufacturers in their first steps towards implementation of eCTD publishing and submission
Hi, Welcome to FWQRC™ Regulatory Focus News Letter
About Blog: FWQRC™ Regulatory focus pharma news, views and analysis of healthcare in a rapidly changing world. Not only do we keep you connected with the latest trends in pharma, we can also help you develop and bring to life your own thoughts, ideas and inspirations to enable you and your business to become key pharma influences
Many of us aware that eCTD is mandatory for DMF submissions from Jan 2020.
- At the beginning of the decision process it is very important to make an evaluation of the current submission processes (“where are we”) in comparison with the eCTD requirements (“where do we need to be”).
- The conclusions drawn from this analysis, together with a careful evaluation of the boundary conditions within the company are the basis for the definition of the User Requirements Specifications (URS).
- In the URS all the needs and boundaries of the expected processes are described and this information is used to find the optimal solution.Three possible solutions are described in detail, with their related advantages and disadvantages
- In-house software,
- Software as a Service (SaaS) and
- Outsourcing
- The last step of the process is the implementation of the chosen solution.
- It has to be considered whether a consultant should help with the creation of this URS document. Especially for the generation of the new processes the experience of a consultant can be helpfulThe URS is part of the official validation documentation according to GAMP and should be established for any new system
- Selection of solution
- Once the URS has been finalised, the most suitable solution has to be found. For this analysis the URS requirements should be classified in some way, e.g. “crucial” and “nice to have”.
- The three possible solutions (In-house Software, Software as a Service and Outsourcing) are described in detail in the following sections. Table 1 compares the most relevant characteristics of the 3 solutions, Table 2: Running an eCTD software system in-house: advantages and disadvantages and Table 3: Host system option: advantages and disadvantages
Table 1: Comparison of the 3 solutions
| Item | In-House software | Software as a Service | Outsourcing |
| Freedom of configuration | High | limited | No |
| Responsibility for update and Maintenance | High | No | No |
| IT support in-house needed | Yes | No | No |
| Link to other IT systems in-house possible | Yes | No | No |
| Initial costs | High | Low | No |
| Ongoing costs | In-house | Yes | Yes |
| Lead time | Long | Medium | Short |
| Scalability | Depends on set-up | Easy | Easy |
| Need of resources and competence for use of eCTD software | Yes | Yes | No |
| confidentiality / data security issues | No | Yes | Yes |
Table 2: Running an eCTD software system in-house: advantages and disadvantages
| Advantages | Disadvantages |
| Full freedom for configuration | High initial costs for setting up the system |
| Free choice of hardware and software components | Relatively long lead time needed to set up the system |
| The software is part of the company-owned software and fits into the IT concept of the company | Full responsibility for update and maintenance |
| Everything stays in house (no data-transfer via internet / confidentiality etc.) | Personnel for technical set-up and maintenance must be available. |
| Link-up to other IT-systems possible (e.g. SAP) | |
| Maintenance costs stay in-house | |
| On-going costs are lower compared to the host software or outsourcing options |
Table 3: Host system option: advantages and disadvantages
| Advantages | Disadvantages |
| Speed: time from the decision to a pilot eCTD is often shorter compared to the in-house software solution | Dependency on an external partner which increases if also the DMS shall be hosted |
| Lower cost for initial implementation as there is no or a smaller initial investment (e.g. initial set-up, user and software licenses, maintenance) | Data transfer via internet (confidentiality, upload / down load capacity) |
| Scalable: ability to scale as business needs change | Data hosted at an external company (confidentiality) |
| No on-going system maintenance | Limited freedom for software configuration |
| On-going costs for renting the system/service |
Table 4: Outsourcing option: advantages and disadvantages
| Advantages | Disadvantages |
| Speed: time from the decision to a pilot eCTD is very short | Dependency on an external partner for each project and throughout the life cycle of a submission |
| No initial investment and no reoccurring costs for system maintenance and technical support | On-going costs for each service during the whole lifecycle of a product/submission (initial submission(s), variations etc.) |
| No direct costs for software, licenses, hardware, system validation and maintenance, training | Data transfer (confidentiality, upload / down load capacity) |
| No need to establish, maintain technical knowledge in building and publishing eCTDs, no need for respective in-house resources (eCTD builder/publisher) | Data hosted at an external company (confidentiality) |
| Scalable: ability to scale as business needs change | |
| Can also substantially-reduce risk of failed initial submissions |
- There are 4 main scenarios that can drive the decision for outsourcing
- there is no in-house software available to build / publish eCTDs or
- the in-house capacities are too little
- to gain experience for the creation of eCTD ready documents and eCTD submissions in-house
- the number of eCTD submissions is too small, seldom use of the system
- Conclusion: The option with the lowest impact on processes and systems in a company is outsourcing. There are various different extents of outsourcing. Common to all is that the external partner will provide the necessary infrastructure / software as well as the personnel to prepare the eCTD.
Stay in connected with FWQRC™ for implementation of eCTD publishing & submission

Merry Christmas to all







HEALTHCARE INSTITUTIONS, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
First FDA-approved vaccine for the prevention of Ebola virus disease
Hi, Welcome to FWQRC Regulatory focus news letter……….
The U.S. Food and Drug Administration announced today the approval of Ervebo, the first FDA-approved vaccine for the prevention of Ebola virus disease (EVD), caused by Zaire ebolavirus in individuals 18 years of age and older.
Cases of EVD are very rare in the U.S., and those that have occurred have been the result of infections acquired by individuals in other countries who then traveled to the U.S., or health care workers who became ill after treating patients with EVD.
“While the risk of Ebola virus disease in the U.S. remains low, the U.S. government remains deeply committed to fighting devastating Ebola outbreaks in Africa, including the current outbreak in the Democratic Republic of the Congo,” said Anna Abram, FDA Deputy Commissioner for Policy, Legislation, and International Affairs.
“Today’s approval is an important step in our continuing efforts to fight Ebola in close coordination with our partners across the U.S. Department of Health and Human Services, as well as our international partners, such as the World Health Organization.
These efforts, including today’s landmark approval, reflect the FDA’s unwavering dedication to leveraging our expertise to facilitate the development and availability of safe and effective medical products to address urgent public health needs and fight infectious diseases, as part of our vital public health mission.”
Thank you for viewing FWQRC blogs……………
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=828&origin=fwqrc.wordpress.com&obj_id=151501655-828-653e3ffa2182fTagged ADE Values, AMV, Analytical Method validation, ANVISA, Audit responses, Auditing, Audits, CAPA, CGMP, Cleaning validation, Dossier preparation, eCTD, EUGMP, Facility registration, GAPAnalysis, NDA labler code, NDMA, Nitrosoamines, PDE Values, Quality Management System, Regulatory Affairs, Risk Assessment, Root cause analysis, Third party audits, USFDA, WHOGMPLeave a comment

FOOD, HEALTHCARE INSTITUTIONS, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
Investigation of Listeria monocytogenes
Welcome to FWQRC Regulatory focus news LetterHere we are going to review on the Outbreak Investigation of Listeria monocytogenes Linked to Hard-Boiled Eggs, December 2019FDA, CDC, and state and local partners are currently investigating a multistate outbreak of Listeria monocytogenes infections linked to foods that contain hard-boiled eggs. On December 20, 2019, Almark Foods recalled and suspended production of hard-boiled and peeled eggs in pails due to the potential for contamination with Listeria monocytogenes. These hard-boiledand peeled eggs were sold in pails under the following names: Rainbow Select Hard-cooked Eggs, Rainbow Select Hard-cooked Eggs in Vinegar, Nic’s Salad Hard-boiled Eggs, Almark Hard-cooked Eggs, and Sutherland Select Hard-cooked Eggs. A full list of recalled products is included below.RecommendationFood processors, restaurants, and retailers should not sell or serve any of the recalled hard-boiled and peeled eggs in pails from Almark Foods. These products were not sold directly to consumers.Additionally, FDA recommends that food processors, restaurants and retailers who have received Almark Foods bulk, fresh hard-boiled eggs, use extra vigilance in cleaning and sanitizing any surfaces that may have come in contact with these products, to reduce the risk of cross-contamination.Background:As of December 17, 2019, a total of seven people infected with the outbreak strain of Listeria monocytogenes have been reported from five states. In interviews, ill people answered questions about the foods they ate and other exposures in the month before they became ill. Of the five people for whom information was available, four reported eating products containing eggs. Three of these people reported eating hard-boiled eggs in deli salads purchased from grocery stores and in salads eaten at restaurants. Illnesses started on dates ranging from April 10, 2017 to November 12, 2019.Additionally, based on whole-genome sequencing, the Listeria monocytogenes found in environmental samples collected at the firm’s processing facility during an FDA inspection conducted in February 2019 is a genetic match to the outbreak strain. FDA is conducting additional inspections and sampling. Almark Foods has been cooperating with the ongoing investigation and announced a voluntary recall of hard-boiled and peeled eggs in pails on December 20, 2019.This outbreak strain was found during environmental sampling in 2017 of one other food facility. That facility is not currently handling food and ceased operation in 2018.Thank you for viewing FWQRC blogs….
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=826&origin=fwqrc.wordpress.com&obj_id=151501655-826-653e40901f7f9Tagged 21CFR, ADE Values, Analytical Method validation, ANVISA, API, Audit responses, Auditing, Audits, CAPA, CDSCO, CGMP, Cleaning validation, CSV, CTD, Digital signature, DMF, DMF deficiencies, Dossier preparation, eCTD, EDQM, Elemental Impurities, EUGMP, Facility registration, FDA, FMEA, GAPAnalysis, GDP, Genotoxicity, GLP, IFRA, IMPURITIES, ISO, ISO standards, Method Development, NDA labler code, NDMA, PDE Values, PhillipinesFDA, Quality Management System, Regulatory Affairs, Risk Assessment, Root cause analysis, ScheduleM, Third party audits, Threshold level, Training, WHOGMPLeave a comment

BIO MEDICAL, HEALTHCARE INSTITUTIONS, LIFE SCIENCES, MEDICAL DEVICES, REGULATORY FOCUS NEWS LETTER
USFDA Class I Recall,the most serious type of recall
Good Morning, Welcome to FWQRC Regulatory Focus News Letter…
Smiths Medical ASD, Inc. Recalls Medfusion® 4000 Syringe Pumps Due to Malfunctioning Alarms and Potential Interruption of Therapy

The FDA has identified this as a Class I recall, the most serious type of recall. Use of these devices may cause serious injuries or death.

Recalled Product
- Medfusion® 4000 Syringe Pump with Firmware Version 1.7.0
- Model numbers 4000-0107-01 and 4000-0106-01
- Manufacturing Dates: June 25, 2019
- Distribution Dates: September 27, 2019 to October 31, 2019
- Devices Recalled in the U.S.: 627
- Date Initiated by Firm: October 28, 2019
- Device Use
The Smiths Medical ASD Medfusion 4000 Syringe Pump is used to deliver blood, blood products or prescribed drugs into a patient’s body in a controlled manner. Syringe pumps are primarily used in the neonatal and pediatric populations and in operating rooms and intensive care units for the adult population.
Reason for Recall
Smiths Medical has become aware of a software issue in the most recently updated Medfusion® 4000 Syringe Pump Firmware, Version 1.7.0, that could potentially cause the low battery alarms to stop working. If the battery alarms do not work, the healthcare provider using the pump will not receive audible or visual notification that the battery is shutting down. This may lead to an interruption of therapy which may lead to serious injury, adverse events, or death.
Smiths Medical has received 74 complaints related to the software update. No injuries or deaths have been reported.
- Who May be Affected
- Healthcare providers using the Smiths Medical ASD, Inc. Medfusion® 4000 Syringe Pump
- Patients who receive therapy delivered by the Smiths Medical ASD, Inc. Medfusion® 4000 Syringe Pump
What to Do
On October 28, 2019, Smiths Medical sent a Recall Notice to customers informing them of the affected models and instructing them to immediately return all affected products.
The Recall Notice from Smiths Medical advised customers to:
- Locate all Medfusion® 4000 Syringe Pumps with Firmware Version 1.7.0 in their possession. The firmware version of the pump can be identified by powering on the unit and observing the firmware version displayed on the screen.
- Determine the number of affected devices in their possession and complete the provided Recall Notice Response Form within 10 days of receipt, returning it to fieldactions@smiths-medical.com even if they do not have any affected product in their possession. All affected product must be returned to Smiths Medical for processing.
- Upon returning the Response Form, Smiths Medical will provide a shipping label to return the affected product. Include a copy of the completed Response Form inside each box of returned product to facilitate processing. Ensure boxes are sealed and labeled with the facility name prior to shipping.
- If they distributed any of the potentially affected products identified in this Recall Notice, they should immediately notify the recipients of the potentially affected products by forwarding them a copy of this Recall Notice.
Thank you for viewing FWQRC blogs…………………….
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=812&origin=fwqrc.wordpress.com&obj_id=151501655-812-653e40902686aTagged 21CFR, ADE Values, AMV, Audit responses, Auditing, Audits, CAPA, CGMP, Digital signature, DMF, DMF deficiencies, eCTD, Elemental Impurities, EUGMP, Facility registration, FMEA, GAPAnalysis, GCP, GDP, GLP, IFRA, ISO standards, Lead Auditor, NDA labler code, NDMA, Nitrosoamines, PDE Values, Quality Management System, Regulatory Affairs, Risk Assessment, Root cause analysis, Training, WHOGMP1 Comment
GUIDELINES, HEALTHCARE INSTITUTIONS, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
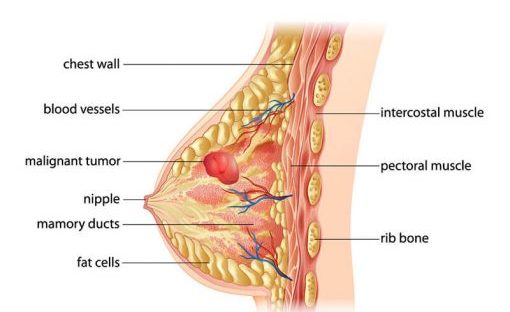
FDA approves new treatment option for patients with HER2-positive breast cancer who have progressed on available therapies
HI, Welcome to FWQRC Regulatory Focus News letter
Today’s topic is about the new treatment option for patients with HER2-positive breast cancer approved by FDA

The U.S. Food and Drug Administration granted accelerated approval to Enhertu (fam-trastuzumab deruxtecan-nxki) for the treatment of adults with unresectable (unable to be removed with surgery) or metastatic (when cancer cells spread to other parts of the body) HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting. Enhertu is a human epidermal growth factor receptor 2 (HER2)-directed antibody and topoisomerase inhibitor conjugate, meaning that the drug targets the changes in HER2 that help the cancer grow, divide and spread, and is linked to a topoisomerise inhibitor, which is a chemical compound that is toxic to cancer cells.

“There have been many advances in the development of drugs for HER2-positive breast cancer since the introduction of Herceptin (trastuzumab) in 1998. The approval of Enhertu represents the newest treatment option for patients who have progressed on available HER2-directed therapies,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research. “Drug development in the area of targeted therapies builds on our scientific understanding of malignant diseases not only in breast cancer, but in multiple other diseases.”
HER2-positive breast cancer is a type of breast cancer that tests positive for a protein called human epidermal growth factor receptor 2 (HER2), which promotes the growth of cancer cells. Approximately one of every five breast cancers have a gene mutation in the cancer cells that makes an excess of the HER2 protein. HER2-positive breast cancers are an aggressive type of breast cancer.

Enhertu’s approval was based on the results of a clinical trial enrolling 184 female patients with HER2-positive, unresectable and/or metastatic breast cancer who had received two or more prior anti-HER2 therapies in the metastatic setting. These patients were heavily pretreated in the metastatic setting, receiving between two and 17 therapies prior to receiving Enhertu. Patients in the clinical trial received Enhertu every three weeks and tumor imagining was obtained every six weeks. The overall response rate was 60.3%, which reflects the percentage of patients that had a certain amount of tumor shrinkage with a median duration of response of 14.8 months.
The prescribing information for Enhertu includes a Boxed Warning to advise health care professionals and patients about the risk of interstitial lung disease (a group of lung conditions that causes scarring of lung tissues) and embryo-fetal toxicity. Interstitial lung disease and pneumonitis (inflammation of lung tissue), including cases resulting in death, have been reported with Enhertu. Health care professionals should monitor for and promptly investigate signs and symptoms including cough, dyspnea (difficult or labored breathing), fever and other new or worsening respiratory symptoms. If these symptoms arise, Enhertu may need to be withheld, the dose reduced or permanently discontinued. Women who are pregnant should not take Enhertu because it may cause harm to a developing fetus or newborn baby, or cause delivery complications. The FDA advises health care professionals to tell females of reproductive age, and males with a female partner of reproductive potential, to use effective contraception during treatment with Enhertu.
The most common side effects for patients taking Enhertu were nausea, fatigue, vomiting, alopecia (hair loss), constipation, decreased appetite, anemia (hemoglobin in blood is below the reference range), decreased neutrophil count (white blood cells that help lead your body’s immune system response to fight infection), diarrhea, leukopenia (other white blood cells that help the immune system), cough and decreased platelet count (component of blood whose function is to react to bleeding from blood vessel injury by clumping, thereby initiating a blood clot). Decreased neutrophil count is a potentially serious and common side effect as described in the Medication Guide. Patients treated with Enhertu may be at increased risk of developing left ventricular dysfunction, which occurs when the heart is unable to pump blood effectively to the body, as this has been seen with other HER2-directed therapies for breast cancer.
Enhertu was granted Accelerated Approval, which enables the FDA to approve drugs for serious conditions to fill an unmet medical need based on a result that is reasonably likely to predict a clinical benefit to patients. Further clinical trials may be required to verify and describe Enhertu’s clinical benefit.
The FDA granted this application Breakthrough Therapy designation, which expedites the development and review of drugs that are intended to treat a serious condition, when preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapies. Enhertu was also granted Fast Track designation, which expedites the review of drugs to treat serious conditions and fill an unmet medical need. This application was approved three months prior to the FDA goal date.
The FDA granted the approval of Enhertu to Daiichi Sankyo.
Thank you for viewing FWQRC blogs……
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=803&origin=fwqrc.wordpress.com&obj_id=151501655-803-653e40902cc21Tagged 21CFR, ADE Values, Analytical Method validation, Auditing, CAPA, Digital signature, Dossier preparation, eCTD, EUGMP, Facility registration, GAPAnalysis, GMP Compliance, IFRA, ISO standards, NDA labler code, Nitrosoamines, PDE Values, Quality Management System, Regulatory Affairs, Risk Assessment, ScheduleM, Training, WHOGMP1 Comment
REGULATORY FOCUS NEWS LETTER, YOUR PARTNER
National Drug Code(NDC)
Hi, Welcome to FWQRC Regulatory focus News Letter….
Here we are going to review the historic step taken by Trump Administration to lower U.S. prescription drug prices
Proposed rule could allow certain prescription drugs to be imported from Canada; draft guidance explains how manufacturers could import drugs, biological products originally intended for sale in another country
Today, President Trump, along with the U.S. Department of Health and Human Services and the U.S. Food and Drug Administration, issued a notice of proposed rulemaking (NPRM) that, if finalized, would allow for the importation of certain prescription drugs from Canada. In addition, the Administration is announcing the availability of a new draft guidance for industry that describes procedures drug manufacturers can follow to facilitate importation of prescription drugs, including biological products, that are FDA-approved, manufactured abroad, authorized for sale in any foreign country, and originally intended for sale in that foreign country.
The NPRM issued today is the first step in implementing a provision of federal law that would allow for the importation of certain prescription drugs from Canada under specific conditions that ensure the importation poses no additional risk to the public’s health and safety while achieving a significant reduction in the cost of covered products to the American consumer. The draft guidance issued today describes procedures for a drug manufacturer to submit documentation that demonstrates that the product offered for import from any foreign country is, in fact, an FDA-approved drug product, including that it is manufactured in accordance with the FDA-approved application.
“Today’s announcement outlines two pathways for the safe importation of certain prescription drugs to help provide safe, effective, more affordable drugs to American patients,” said Health and Human Services Secretary Alex Azar. “These are historic actions by HHS and the FDA, and they represent the bold nature of President Trump’s agenda for lowering drug costs. The President has recognized the opportunity to lower costs for American patients through safe importation, and we at HHS and FDA are delivering on that possibility through a safe, commonsense approach.”
The NPRM would allow states and certain other non-federal government entities to submit importation program proposals to the FDA for review and authorization. An importation program could be co-sponsored by a pharmacist, a wholesaler, or another state or non-federal governmental entity. Referred to as Section 804 Importation Programs, these programs would be authorized by the FDA to manage the importation of certain prescription drugs that are approved in Canada and also meet the conditions in an FDA-approved drug application. Eligible prescription drugs would have to be relabeled with the required U.S. labeling prior to importation and undergo testing for authenticity, degradation, and to ensure that the drugs meet established specifications and standards. Notably, these programs would also have to demonstrate significant cost reductions to the American consumer.
“The FDA continues to assess and act on multiple opportunities to promote competition that can, in turn, help reduce drug prices and improve access to medicines for Americans,” said Assistant Secretary for Health Brett Giroir. “The proposed rule and draft guidance include procedures intended to protect the public’s health and safety. We look forward to receiving public comment on these draft policies, and we will take timely comments into account as we work to finalize the rule and guidance. Our ultimate goal is to provide a robust program that clearly lays out procedures to import drugs that could provide lower prices while also maintaining the high quality Americans expect.”
Of note, the draft guidance describes procedures drug manufacturers could follow to obtain an additional National Drug Code (NDC) for certain FDA-approved prescription drugs, including biological products, that were originally manufactured, and intended to be marketed, in a foreign country. The use of an additional NDC would allow greater flexibility for drug companies to offer these products at a lower price than what their current distribution contracts require.
The draft guidance also recommends that the drug manufacturer include a statement on the product’s label and in the prescribing information to assist pharmacists to accurately identify, dispense and bill for these products. Prescription drugs, including biological products, imported under the pathway described in the draft guidance could be available to patients in a variety of settings, including hospitals, health care providers’ offices, or licensed U.S. pharmacies, and would include the FDA-approved labeling (including prescribing information).
Comments on the NPRM are being accepted for 75 days after publication in the Federal Register and comments on the draft guidance are being accepted for 60 days after publication in the Federal Register
Thank you for viewing FWQRC newsletters……..
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=800&origin=fwqrc.wordpress.com&obj_id=151501655-800-653e40903041eTagged ADE Values, Analytical Method validation, ANVISA, API, Audit responses, Auditing, Audits, CAPA, CDSCO, CGMP, Cleaning validation, CSV, CTD, DMF, DMF deficiencies, Dossier preparation, eCTD, EDQM, Facility registration, FDA, Genotoxicity, GMP, GMP Compliance, IFRA, IMPURITIES, ISO, ISO standards, Lead Auditor, Method Development, MOH, NDA labler code, NDMA, PDE Values

Some Medicines and Driving Don’t Mix
Hi, Welcome to FWQRC News Letter
Here we are going to review on medicines that might affect driving
Although most medications won’t affect your ability to drive, some prescription and nonprescription medicines can have side effects and cause reactions that may make it unsafe to drive, including:
- sleepiness/drowsiness
- blurred vision
- dizziness
- slowed movement
- fainting
- inability to focus or pay attention
- nausea
- excitability
Additionally, we know that CBD can cause sleepiness, sedation and lethargy, based on data from the approved prescription CBD drug. Because of these side effects, consumers should use caution if planning on operating a motor vehicle after consuming any CBD products.
Medicines That Might Affect Driving
Knowing how your medications — or any combination of them — affect your ability to drive is a safety measure. Some drugs that could make it dangerous to drive include:
- opioid pain relievers
- prescription drugs for anxiety (for example, benzodiazepines)
- anti-seizure drugs (antiepileptic drugs)
- antipsychotic drugs
- some antidepressants
- products containing codeine
- some cold remedies and allergy products, such as antihistamines (both prescription and OTC)
- sleeping pills
- muscle relaxants
- medicines that treat or control symptoms of diarrhea
- medicines that treat or prevent symptoms of motion sickness
- diet pills, “stay awake” drugs, and other medications with stimulants (e.g., caffeine, ephedrine, pseudoephedrine)
- Taking Cannabidiol (CBD) Products and Driving Can Be Dangerous
Last year the agency approved a prescription CBD drug, Epidiolex, for the treatment of two rare and life-threatening seizure disorders in children. The FDA has not approved any other CBD products. Unlike drug products approved by the FDA, unapproved CBD drug products have not been subject to any FDA review.
Moreover, there has been no FDA evaluation regarding whether unapproved CBD drug products are safe and effective to treat a particular disease or condition, what the proper dosage is, how they could interact with other drugs, foods or cosmetics, or whether they have dangerous side effects or other safety concerns.
But we do know that CBD can cause sleepiness, sedation and lethargy, based on data from the approved prescription drug. Because of these side effects, consumers should use caution if planning on operating a motor vehicle after consuming any CBD products.
We urge you to obtain more information about CBD by visiting: What You Need to Know About Products Containing Cannabis or Cannabis-derived Compounds, Including CBD.
Some Sleep Medicines Can Impair You, Even the Next Morning
People with insomnia have trouble falling or staying asleep. Many take medicines to help sleep. Come morning, though, some sleep medicines could make you less able to perform activities for which you must be fully alert, including driving.
A common ingredient in a widely prescribed sleep medication is zolpidem, which belongs to a class of medications called sedative-hypnotics. The FDA has found that medicines containing zolpidem, especially extended release forms, can impair driving ability and other activities the next morning.
Zolpidem immediate and extended-release forms are marketed as generic drugs and under these brand names:
- Ambien and Ambien CR (oral tablet)
- Edluar (tablet placed under the tongue)
- Intermezzo (tablet placed under the tongue)
- Zolpimist (oral spray)
People who take sleep medicines should talk to their health care professional about ways to take the lowest effective dose. Don’t assume that non-prescription sleep medicines are necessarily safer alternatives. The FDA is also evaluating the risk of next-day impairment with other insomnia drugs, both prescription and OTC versions.
Allergy Medicines Can Affect Your Ability to Drive
For allergy sufferers, medications containing antihistamines can help relieve many different types of allergies, including hay fever. But these medicines may interfere with driving and operating heavy machinery (including driving a car).External Link Disclaimer Antihistamines can slow your reaction time, make it hard to focus or think clearly, and may cause mild confusion even if you don’t feel drowsy.
Read the OTC Drug Facts label of your medicine and understand the warnings before using it. Also, avoid drinking alcohol or taking sleep medications while using some antihistamines. Those combinations can increase the sedative effects of antihistamines.
How to Avoid Driving Impaired
You can still drive safely while taking most medications. Talk to your health care provider about possible side effects. For example, some antihistamines and sleep medications work for longer periods than others. You might feel the sedating effects of these medications for some time after you’ve taken them, and maybe even into the next day.
Doctors and pharmacists can tell you about known side effects of medications, including those that interfere with driving. You can also request printed information about the side effects of any new medicine.
To manage or minimize side effects while driving, your health care provider may be able to adjust your dose, adjust the timing of when you take the medicine, or change the medicine to one that causes fewer side effects for you.
Here are some more tips:
- Always follow directions for use and read warnings on medication packaging, or handouts provided by the pharmacy.
- Don’t stop using your medicine unless your prescriber tells you to.
- Tell your health care provider about all the products you are taking, including prescription, OTC, and herbal products. Also, let them know about any reactions you experience.
Thank you for visiting FWQRC blogs…
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=792&origin=fwqrc.wordpress.com&obj_id=151501655-792-653e409034a11Tagged ADE Values, Analytical Method validation, ANVISA, Auditing, Audits, CAPA, CDSCO, Cleaning validation, CSV, DMF, eCTD, EDQM, Facility registration, GMP Compliance, IFRA, Lead Auditor, MOH, NDA labler code, NDMA, PDE Values, PhillipinesFDA, Projects, Quality Management System, Regulatory Affairs, Risk Assessment, USFDA, WHOGMP1 Comment

5 Tips to Consider When Preparing Recommendations for Changes to Retail Food Policy
Hi, Welcome to FWQRC Regulatory Focus News letter
Developing recommended changes for the most effective, efficient, and feasible policy is challenging. Through our work in retail food policy analysis, we have identified five things individuals can consider to prepare retail food policy recommendations.
1. Define the problem that needs to be addressed
- Describe and provide evidence for the existence, size, and severity of the problem.
- Public policy making is about problem solving. If no problem exists, there is no need to offer a policy solution. Establishing policy to fix a nonexistent problem may unduly burden stakeholders.
2. Describe the cause of the problem
- Provide the reasoning and evidence to support a link between the problem and cause.
- Problem solving requires identifying, understanding, and explaining the underlying cause(s) of the problem. Providing information about the source of the problem will help to choose the best solution. Anticipate that your reasoning and evidence may be challenged.
3. Explain why the current policy is not addressing the problem
- Explain what barriers or challenges exist that make the current policy insufficient.
- The existing policy may not be sufficient to solve the problem. There may be formal challenges such as existing laws, and informal challenges such as differing perceptions or lack of awareness or training that you should consider and explain.
4. Present your policy recommendation and explain how it compares to possible alternatives
- Provide information on alternative solutions considered and explain how and why your policy recommendation is preferable.
- Policy recommendations should offer a solution to the problem that was identified.
- Sometimes policy isn’t the best solution to a problem. Gaps in knowledge, resources, and enforcement of current policy can impact effectiveness. Before proposing a new policy or changes to policy, alternatives should be considered. This should include a description of the criteria you used for the comparison.
5. Characterize the impact of your proposed policy recommendation
- Describe the intended and/or unintended consequences, positive and negative, that may result from implementing the proposed policy recommendation
- Do the positive consequences outweigh the negative consequences? Describe why the proposal is a more effective, efficient, and feasible solution.
- Anticipate stakeholder questions and concerns
- Be prepared to address questions and concerns regarding areas of potential disagreement.
Questions may come in the form of:
A fact – what is and what isn’t or what happened or didn’t happen
A value – what is appropriate and inappropriate
Or a policy – what we should do or what the policy should be
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=780&origin=fwqrc.wordpress.com&obj_id=151501655-780-653e413796d7b1 Comment
LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
New analysis highlights link between generic drug competition and lower drug prices, underscores importance of FDA efforts to spur generic drug development and market entry
Hi, Welcome to FWQRC Regulatory Focus News letter
- The U.S. Food and Drug Administration posted a new analysis showing greater competition among generic drug makers is associated with lower generic drug prices.

- It updates and expands on a previous analysis from 2005 and includes two different sources of wholesale drug prices – average manufacturer prices (AMP) reported to the Centers for Medicare and Medicaid Services and invoice-based wholesale prices reflecting pharmacy acquisitions from IQVIA’s National Sales Perspective database.
- The analysis looked at all drug products that had initial generic entry between 2015 and 2017 and showed that as competition increases, generic drug prices decline.
- For products with a single generic producer, the generic AMP is 39% lower than the brand drug price before generic competition, compared to a 31% reduction using invoice-based drug prices.
- With two competitors, AMP data show that generic prices are 54% lower than the brand drug price before generic competition, compared to a 44% reduction using invoice-based drug prices.
- With four competitors, AMP data show that the generic prices are 79% less than the brand drug price before generic entry, compared to a 73% reduction using invoice-based drug prices.

- With six or more competitors, generic prices using both AMP and invoice-based drug prices show price reductions of more than 95% compared to brand prices before generic entry.
- This analysis builds on earlier FDA work External Link Disclaimer comparing generic and brand drug prices and follows related studies.
- Through ongoing efforts under the FDA’s Drug Competition Action Plan and the Generic Drug User Fee Amendments, the agency is further encouraging robust and timely market competition for generic drugs and helping bring greater efficiency and transparency to the generic drug review process, without sacrificing the scientific rigor underlying our generic drug program.
- “This new analysis underscores the critical nature of the work the FDA is undertaking to encourage timely market competition from high-quality, safe and effective generic drugs, which can help provide patients with access to affordable therapies that treat a wide range of medical conditions.
- “The FDA will continue to do all it can to support a robust, competitive generic drug market by helping facilitate a strong pipeline of applications, improving agency processes and providing industry with thoughtful, clear guidance and recommendations to support generic drug development and approval.”
Thank you for visiting FWQRC blogs
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=773&origin=fwqrc.wordpress.com&obj_id=151501655-773-653e41379b30c1 Comment

GMP, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER, RULES
FDA Finalizes Rule to Go From Paper to Electronic Devices Submissions
Hi, Welcome to FWQRC Regulatory Focus News Letter.
Here we are going to discuss about the finalised rule for the submission process.
As part of an effort to improve the US Food and Drug Administration’s (FDA) medical device submission process, the agency on Friday issued a final rule to remove the requirements for multiple paper copy submissions and replace them with a single electronic submission.
The agency said the rule, proposed in September 2018 and will take effect in 30 days, is in response to an executive order from the Trump Administration in 2017 made famous as the “one-in, two-out” order. FDA’s rule is meant to improve the device premarket submission program and create a more efficient submission system.
“The requirement for a single submission in electronic format applies to all submission types that fall within the provisions listed in section 745A(b) of the FD&C Act; under this final rule, FDA is only amending those regulations that specifically mention paper and/or multiple copies of such regulatory submissions and are not consistent with this final rule,” FDA said.
The agency responded to four comments on the proposal but did not update the rulemaking. The agency noted that the final rule will produce cost savings for firms without imposing any additional regulatory burdens for submissions or affect the agency’s ability to review submissions.
“Firms will incur minimal administrative costs to read and understand the rule. We expect the economic impact of this regulation to be a total net costs savings yielding positive net benefits,” FDA said.
The agency also noted that submissions in electronic format can include those created and submitted on CD, DVD or flash drive and mailed to FDA.
For premarket approval applications (PMAs), the final rule changes one section to take out a section requiring an applicant to submit three copies of any updated safety and effectiveness report for pending applications.
FDA also previously required that PMAs be submitted in six copies, each bound in one or more numbered volumes, but that language has been removed with this final rule.
In another section, FDA removes the requirement that a PMA applicant has to provide copies of information that it believes to be trade secret or confidential commercial or financial information in the PMA.
FDA in September also published a draft guidance with “both binding and nonbinding provisions”
Please contact FWQRC for electronic submissions. (+91 8072483812)
Share this:
Loading…Tagged Analytical Method validation, API, Audit responses, Auditing, CAPA, CGMP, CTD, DMF, DMF deficiencies, Dossier preparation, eCTD, EDQM, FDA, Genetics, Genotoxicity, ISO standards, Lead Auditor, Method Development, NDMA, Nitrosoamines, PHARMACEUTICAL MANUFACTUERES, Quality Management System, Regulatory Affairs, Risk Assessment, Third party audits, Threshold level, USFDALeave a comment

MEDICAL DEVICES, REGULATORY FOCUS NEWS LETTER
Evaluation of Automatic Class III Designation (De Novo) Summaries
Hi, Welcome to FWQRC Regulatory Focus News Letter

Here we are going to discuss about the Food and Drug Administration Modernization Act of 1997 (FDAMA) added the De Novo classification option as an alternate pathway to classify novel medical devices that had automatically been placed in Class III after receiving a “not substantially equivalent” (NSE) determination in response to a premarket notification [510(k)] submission. Section 513(f)(2) of the FD&C Act was amended by section 607 of the Food and Drug Administration Safety and Innovation Act (FDASIA), on July 9, 2012, to allow a sponsor to submit a De Novo classification request to the FDA for without first being required to submit a 510(k).
There are two options for De Novo classification for novel devices of low to moderate risk.
Option 1: Any person who receives an NSE determination in response to a 510(k) submission may, within 30 days of receipt of the NSE determination, submit a De Novo request for the FDA to make a risk-based evaluation for classification of the device into Class I or II.
Option 2: Any person who determines that there is no legally marketed device upon which to base a determination of substantial equivalence may submit a De Novo request for the FDA to make a risk-based classification of the device into Class I or II, without first submitting a 510(k) and receiving an NSE determination.
Devices that are classified through the de novo process may be marketed and used as predicates for future 510(k) submissions.
Since 2010, the FDA has begun releasing summary documents for devices classified through the De Novo process. The De Novo summary is intended to present an objective and balanced summary of the scientific evidence that served as the basis for the decision to grant a De Novo request. The De Novo summary also serves as a resource regarding the types of information necessary to support substantial equivalence for device manufacturers that may wish to use the device as a predicate for future 510(k) submissions.
Thank you for visiting FWQRC blogs…
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
Loading…Leave a comment

GMP, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
Managing Food Safety: The HACCP approach for food safety
Hi, Welcome to FWQRC Regulatory Focus news letter
Here we are going to discuss about the manual for the Voluntary Use of HACCP Principles for Operators of Food Service and Retail Establishments

FDA endorses the voluntary implementation of food safety management systems in retail and food service establishments. Combined with good basic sanitation, a solid employee training program, and other prerequisite programs, HACCP can provide you and your employees a complete food safety management system.

The goal in applying HACCP principles in retail and food service is to have you, the operator, take purposeful actions to ensure safe food. You and your regulatory authority have a common objective in mind – providing safe, quality food to consumers. Your health inspector can help you achieve this common objective, but remember that the ultimate responsibility for food safety at the retail level lies with you and your ability to develop and maintain an effective food safety management system.
Managing food safety should be as fully integrated into your operation as those actions that you might take to open in the morning, ensure a profit, or manage cash flow. By putting in place an active, ongoing system, made up of actions intended to create the desired outcome, you can achieve your goal of improving food safety. The application of the HACCP principles provides one system that can help you accomplish that goal.
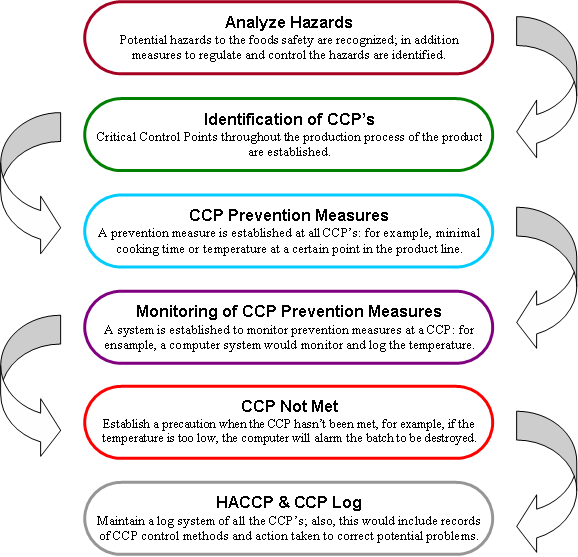
This Manual will provide details on how to organize your products so that you can voluntarily develop your own food safety management system using HACCP principles. The HACCP plans that you will develop using this Manual, in combination with prerequisite programs (discussed in Chapter 3), will constitute a complete food safety management system. Partnering with your regulatory authority or other food safety professional is recommended, but the design, implementation, and success of your system rests with you.
Thank you for visiting FWQRC blogs…..
Contact FWQRC for HACCP implementation (fwqrcservices@gmail.com) (+91 8072483812)
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=752&origin=fwqrc.wordpress.com&obj_id=151501655-752-653e4137a73dcLeave a comment

LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
Drug Study Design

- Before a new drug or biologic can be marketed, its sponsor must show, through adequate and well-controlled clinical studies, that it is effective. A well-controlled study permits a comparison of subjects treated with the new agent with a suitable control population, so that the effect of the new agent can be determined and distinguished from other influences, such as spontaneous change, “placebo” effects, concomitant therapy, or observer expectations. FDA regulations [21 CFR 314.126] cite five different kinds of controls that can be useful in particular circumstances:
- placebo concurrent control
- dose-comparison concurrent control
- no-treatment concurrent control
- active-treatment concurrent control, and
- historical control
- No general preference is expressed for any one type, but the study design chosen must be adequate to the task. Thus, in discussing historical controls, the regulation notes that, because it is relatively difficult to be sure that historical control groups are comparable to the treated subjects with respect to variables that could effect outcome, use of historical control studies has been reserved for special circumstances, notably cases where the disease treated has high and predictable mortality (a large difference from this usual course would be easy to detect) and those in which the effect is self-evident (e.g., a general anesthetic).
Placebo control, no-treatment control (suitable where objective measurements are felt to make blinding unnecessary), and dose-comparison control studies are all study designs in which a difference is intended to be shown between the test article and some control. The alternative study design generally proposed to these kinds of studies is an active-treatment concurrent control in which a finding of no difference between the test article and the recognized effective agent (active-control) would be considered evidence of effectiveness of the new agent. There are circumstances in which this is a fully valid design. Active-controls are usually used in antibiotic trials, for example, because it is easy to tell the difference between antibiotics that have the expected effect on specific infections and those that do not. In many cases, however, the active-control design may be simply incapable of allowing any conclusion as to whether or not the test article is having an effect.
There are three principal difficulties in interpreting active-control trials. First, active-control trials are often too small to show that a clinically meaningful difference between the two treatments, if present, could have been detected with reasonable assurance; i.e., the trials have a high “beta-error.” In part, this can be overcome by increasing sample size, but two other problems remain even if studies are large. One problem is that there are numerous ways of conducting a study that can obscure differences between treatments, such as poor diagnostic criteria, poor methods of measurement, poor compliance, medication errors, or poor training of observers. As a general statement, carelessness of all kinds will tend to obscure differences between treatments. Where the objective of a study is to show a difference, investigators have powerful stimuli toward assuring study excellence. Active-control studies, however, which are intended to show no significant difference between treatments, do not provide the same incentives toward study excellence, and it is difficult to detect or assess the kinds of poor study quality that can arise. The other problem is that a finding of no difference between a test article and an effective treatment may not be meaningful. Even where all the incentives toward study excellence are present, i.e., in placebo-controlled trials, effective drugs are not necessarily demonstrably effective (i.e., superior to placebo) every time they are studied. In the absence of a placebo group, a finding of no difference in an active-control study therefore can mean that both agents are effective, that neither agent was effective in that study, or that the study was simply unable to tell effective from ineffective agents. In other words, to draw the conclusion that the test article was effective, one has to know with assurance that the active-control would have shown superior results to a placebo, had a placebo group been included in the study.
For certain drug classes, such as analgesics, antidepressants or antianxiety drugs, failure to show superiority to placebo in a given study is common. This is also often seen with antihypertensives, anti-angina drugs, anti-heart failure treatments, antihistamines, and drugs for asthma prophylaxis. In these situations, active-control trials showing no difference between the new drug and control are of little value as primary evidence of effectiveness and the active-control design (the study design most often proposed as an alternative to use of a placebo) is not credible.
In many situations, deciding whether an active-control design is likely to be a useful basis for providing data for marketing approval is a matter of judgment influenced by available evidence. If, for example, examination of prior studies of a proposed active-control reveals that the test article can very regularly (almost always) be distinguished from placebo in a particular setting (subject population, dose, and other defined parameters), an active-control design may be reasonable if it reproduces the setting in which the active-control has been regularly effective.
It is often possible to design a successful placebo-controlled trial that does not cause investigator discomfort nor raise ethical issues. Treatment periods can be kept short; early “escape” mechanisms can be built into the study so that subjects will not undergo prolonged placebo-treatment if they are not doing well. In some cases randomized placebo-controlled therapy withdrawal studies have been used to minimize exposure to placebo or unsuccessful therapy; in such studies apparent responders to a treatment in an open study are randomly assigned to continued treatment or to placebo. Subjects who fail (e.g., blood pressure rises, angina worsens) can be removed promptly, with such failure representing a study endpoint.
IRBs may face difficult issues in deciding on the acceptability of placebo-controlled and active-control trials. Placebo-controlled trials, regardless of any advantages in interpretation of results, are obviously not ethically acceptable where existing treatment is life-prolonging. A placebo-controlled study that exposes subjects to a documented serious risk is not acceptable, but it is critical to review the evidence that harm would result from denial of active treatment, because alternative study designs, especially active-control studies, may not be informative, exposing subjects to risk but without being able to collect useful information.
Thank you for visiting FWQRC blogs……
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
Loading…Leave a comment

GMP, HEALTHCARE INSTITUTIONS, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
FDA underscores that consumers should not use drugs, dietary supplements and devices recalled from Basic Reset and Biogenyx following consent decree for federal violations
Hi, Welcome to FWQRC Regulatory Focus News letter……
Here we are going to discuss about the recent drug alert
The U.S. Food and Drug Administration is alerting consumers of a recall of 25 drug, dietary supplement and medical device product lines distributed by Basic Reset and Biogenyx of Hendersonville, Tennessee. In September, a federal court entered a consent decree of permanent injunction between the United States and the two companies and their owner, Fred R. Kaufman III. Under the consent decree, Basic Reset and Biogenyx must recall and stop distributing products until the companies comply with the Federal Food, Drug, and Cosmetic Act and other requirements listed in the consent decree.
Basic Reset and Biogenyx products that have been recalled include drugs such as Earth Wash and Ionyte, as well as dietary supplements Mello-Tonin and Body Mass Reset and device Energy FX among others. Basic Reset and Biogenyx issued recall notices by email to their customers on Sept. 23 and Oct.18, requesting disposal or return to the place of purchase for products sold, purchased or distributed after Nov. 7, 2017. Given these products do not comply with appropriate FDA standards, they have the potential to be unsafe or ineffective for their particular uses, and could lead to adverse health impacts. The FDA is reminding consumers who may still have these products not to use them and distributors not to sell any of the recalled products as they do not meet FDA regulations.
“The FDA’s laws are designed to protect the public health by ensuring, among other things, that drugs and medical devices are safe and effective for their intended uses and that dietary supplements are manufactured and distributed appropriately,” said Melinda K. Plaisier, FDA Associate Commissioner for Regulatory Affairs. “All companies must follow the appropriate standards and are given the opportunity to ensure their actions are in accordance with these laws. We will continue to prevent the distribution of products that do not comply with applicable FDA requirements and ultimately place the public health at risk.”
Basic Reset and Biogenyx have not received the FDA’s approval for the sale of their drugs and one device, despite the companies’ claims that these products can be used to diagnose, cure, mitigate, treat or prevent conditions such as inflammation, chronic diarrhea, bacterial infections, head lice, allergies and pain. Consumer use of an unapproved product that claims to treat diseases may cause them to delay seeking appropriate medical care. Additionally, unapproved products have not been reviewed by the FDA for quality, safety or effectiveness. Basic Reset and Biogenyx also unlawfully distributed dietary supplements that are adulterated and misbranded.
Thanks for visiting FWQRC blogs…..
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
Loading…Leave a comment

HEALTHCARE INSTITUTIONS, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER, RULES
Final Rule to Collect Antimicrobial Sales and Distribution Information by Animal Species
Hi, Welcome to FWQRC Regulatory Focus News Letter
Here we are going to discuss about the final rule to collect Antimicrobial Sales and Distribution information by Animal Species

The FDA has issued a final rule to obtain more detailed information about antimicrobials sold or distributed for use in food-producing animals by including estimates of sales data by species. The additional data will improve understanding about the extent to which antimicrobials are sold or distributed for use in major food-producing species and help the FDA further target its efforts to ensure judicious use of medically important antimicrobials. It will also assist the agency in measuring the effectiveness of those efforts.
The final rule requires animal drug sponsors to submit species-specific estimates of antimicrobial sales for cattle, swine, chickens, and turkeys. The final rule also includes a provision to improve the timeliness of FDA’s annual summary report of these sales data by requiring the FDA to publish its annual summary report of antimicrobial sales and distribution information by December 31 of the following year.
Thank you for visiting FWQRC blogs…..
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
Loading…Leave a comment

HEALTHCARE INSTITUTIONS, LIFE SCIENCES, PHARMA, REGULATORY FOCUS NEWS LETTER
BIOSIMILAR DRUGS
Good Morning, Welcome to FWQRC Regulatory Focus News Letter
Today We are going to review the benefits and concerns on biosimilar drugs

Pathway for approvals was initiated in 2009.
Ten years later (or less than five years since the first FDA approval of a biosimilar), and just 42% (11 out of 26) of FDA-approved biosimilars have launched. But in the next three months , a clutch of new biosimilars will hit the market, including new ones in oncology, hinting at a wave of uptake.

For instance, Pfizer is expected to launch three biosimilars soon: one for Avastin (bevacizumab) later this month, one for Rituxan (rituximab) next month, and one in February for Herceptin (trastuzumab). Two other trastuzumab biosimilars may also launch soon, which would mean more than 60% of biosimilars approved in the US will have launched by early next year.
The rising number of launches, combined with an increasing amount of quick uptake, may put biosimilar foes on their heels.
For instance, Neulasta (pegfilgrastim) biosimilars have found recent success, with Coherus’ Udenyca (pegfilgrastim-cbqv) and Mylan and Biocon’s Fulphila (pegfilgrastim-jmdb) capturing 25% market share in just over a year, according to a report released last week from Bernstein.
Similarly, a sign of rapid uptake can be seen with Amgen’s Mvasi (bevacizumab-awwb), which has captured 10% of the Avastin market in just four months.
“Biosimilars are growing their market share and leading to meaningful price erosion over time; with the more recent biosimilar launches showing a lot of success – reflecting perhaps the growing market sophistication of the biosimilar companies,” former FDA Commissioner Scott Gottlieb, referring to the Bernstein report, noted recently.
And in the future, Humira (adalimumab) and Enbrel (etanercept) biosimilars (seven approved, zero launched int he US) may look more like outliers in a larger pool of approvals and subsequent launches. By contrast, in the EU, Humira biosimilars have already captured 35% of the multi-billion-dollar market in one year, and biosimilars have captured 50% of the Enbrel market in about three years, according to Bernstein.
The US Remicade (infliximab) biosimilar market is also an eyesore (Bernstein refers to it as “essentially a failed market”) as the two biosimilar entrants have only amassed 12% market share in more than two years. Amgen’s infliximab biosimilar was recently approved last week and may hit the market soon. And Johnson & Johnson said the Federal Trade Commission has launched an investigation into its contracting practices for Remicade, although similar investigations in Canada and the UK yielded little.
Thank you for visiting FWQRC blogs…..
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
Loading…Leave a comment

INSPECTION, REGULATORY FOCUS NEWS LETTER
FDA Provides Flexibility for Start of Routine Inspections on Small Farms
Hi, Welcome to FWQRC Regulatory Focus News Letter
Here we are going to discuss about flexibility for Start of Routine Inspection on small farms
The U.S. Food and Drug Administration is providing flexibility for when states may begin conducting routine inspections of small farms, other than sprouts operations, under the Food Safety Modernization Act (FSMA) Produce Safety Rule.

Routine inspections of small farms, other than sprouts operations, subject to the Produce Safety Rule, will generally begin in Spring 2020; however, the FDA is clarifying that states receiving competition A/B funding as part of the State Produce Implementation Cooperative Agreement Program (CAP) may begin routine inspections as early as January 1, 2020. This clarification is being made after several requests from states to have greater flexibility to align routine inspections with the winter growing season where applicable. Individual states will make final decisions on whether to initiate their first routine inspections of small farms at the earlier date in January 2020. States that want to begin routine inspections of small farms, other than sprouts operations, on January 1, 2020, should prioritize completing their planned inspections of large farms subject to the rule before conducting routine inspections of those small farms.
The major compliance date for small farms, other than sprouts operations, subject to the Produce Safety Rule arrived on January 28, 2019; however, FDA had previously announced that routine inspections would not begin until Spring 2020. The delayed start to routine inspection follows a similar delay for the start of routine inspections of large farms (other than sprout operations), both done to provide the FDA and its state partners additional time to conduct education and outreach.
On Farm Readiness Reviews (OFRR), offered in collaboration with the National Association of State Departments of Agriculture (NASDA), will continue to serve as a helpful tool to farmers as they determine how prepared they are to comply with the requirements of the Produce Safety Rule.
Additional information about inspections, the rule and any related resources can be found on the Produce Inspections webpage at FDA.gov.
Thanks for visiting FWQRC blogs……
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=684&origin=fwqrc.wordpress.com&obj_id=151501655-684-653e4208eb406Leave a comment

GMP, LIFE SCIENCES, PHARMA, REGULATORY FOCUS NEWS LETTER
MUTUAL RECOGNITION AGREEMENT (MRA) FROM A GLOBAL PERSPECTIVE
Hi, Welcome to FWQRC Regulatory Focus New Letter
Here we are going to measuring the Impact of the Pharmaceutical Annex to the U.S./EU Mutual Recognition Agreement

When we first began discussing the prospects of a mutual recognition agreement with the European Union, in which EU member state (EUMS) regulators and the FDA would agree to rely on each other’s factual findings from their good manufacturing practice (GMP) inspections of drug facilities, the potential benefits seemed obvious.
European drug makers to benefits from MRA
By relying on each other’s expertise and ability to conduct GMP inspections under the Pharmaceutical Annex to the U.S./EU Mutual Recognition Agreement, we expected to avoid duplicate inspections of the same facilities, especially for facilities with a strong record of compliance. And these efficiencies would allow us to reallocate our inspectional resources to areas of higher risk. Of course, we knew we would have to wait until the agreement was implemented before we could confirm these assumptions.
Developing the MRA took time and resources. Beginning in 2014, FDA experts were sent to Europe to observe EU officials audit each of the 28 EUMS inspectorates to assure that they could conduct inspections at a standard similar to a U.S. inspection. In addition, we had to establish confidentiality commitments with each regulator to allow for the sharing of non-public information contained in inspection reports and be assured that each EUMS had a framework for preventing conflicts of interest that provided a similar level of protection as those in place at FDA. The FDA completed this work for the most common type of drug manufacturing inspections in July 2019.
The FDA and the EU have been collecting data on the operational impact of the MRA ever since the first countries were found capable on November 1, 2017, and the numbers are quite promising. So far the EU has conducted 29 inspections at FDA’s request and the FDA has conducted 14 inspections at the EU’s request. Moreover, FDA has deferred 157 inspections in the EU after review of the inspectional information provided by our trusted partners. We anticipate seeing an even greater impact now that the MRA has been implemented in all 28 EU countries for human drug products.
The need for such metrics was one of the recommendations from the National Academies of Sciences, Engineering, and Medicine (NASEM) in a recent report. FDA had asked an Ad Hoc NASEM committee to examine mutual recognition and mutual reliance agreements around the world (some dating back many years) and the committee found a surprising lack of data on the successes and challenges of these programs. To address that information gap, the committee called on regulatory authorities to create a results framework with clear indicators, metrics, and processes for monitoring and evaluating these programs. Such information would increase understanding of the program’s public health benefits and enable benefit-risk analyses over time, the report’s authors said.
We certainly agree that more data is needed on the U.S./EU MRA, beyond the operational data we’re collecting now. In fact, we’re working with our EU partners to explore what additional metrics might be informative. Having sufficient data will help the FDA decide not only how to allocate our inspectional resources, but also the scope and breadth of future mutual recognition and mutual reliance agreements that might include other categories of inspections and product types, such as veterinary medicines. Since these other categories often involve different inspectorates in Europe than those audited for human pharmaceuticals, implementation could take some time. Yet, eventually, with metrics in place, we could confirm their benefits as well, allowing manufacturers to avoid unneeded inspections while ensuring that medicines are safer for patients.
Thank you for visiting FWQRC blogs
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
Loading…Leave a comment
BIO MEDICAL, HEALTHCARE INSTITUTIONS, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
FDA Issues Draft Guidance on Performance Criteria for Magnetic Resonance (MR) Coils
Hi, Welcome to FWQRC Regulatory Focus News letter
Greetings from FWQRC………..
Here we are going to discuss about the recent draft guidance on performance criteria for MR Coils
Facts about the draft guidance
This draft guidance is intended to provide performance criteria for magnetic resonance (MR) coils in support of the Safety and Performance Based Pathway.
Under the Safety and Performance Based Pathway, medical device manufacturers planning to submit a 510(k) will have the option to use the performance criteria as identified in a final guidance to support substantial equivalence, rather than direct comparison of the performance of the subject device to that of a predicate device.
What is MR Coils



Framework for the Safety and Performance Based Pathway
Overview
There are three types of Premarket Notification 510(k)s that may be submitted to FDA: Traditional; Special; and Abbreviated. The Special and Abbreviated 510(k) programs were developed in 1998 and described in the “New 510(k) Paradigm” to facilitate the 510(k) review process for certain types of submissions subject to 510(k) requirements. In 2019, the FDA split “The New 510(k) Paradigm” into separate guidance documents; The Special 510(k) Program and The Abbreviated 510(k) Program. The Safety and Performance Based Pathway is an expansion of the concept of the Abbreviated 510(k) pathway for certain, well understood device types.
The FDA expects to operationalize this pathway once the first device types and applicable performance criteria have been identified and final guidances have been published. Once the FDA begins to operationalize this pathway, a medical device manufacturer will have the option to meet FDA-identified performance criteria to demonstrate that its device is as safe and effective as a predicate device. The use of this pathway does not affect the FDA’s ability to request any information authorized by the statute or regulations.
What device types are appropriate for the Safety and Performance Pathway?
The Safety and Performance Based Pathway is appropriate when FDA has determined that:
The new device has the same indications for use as, and technological characteristics that do not raise different questions of safety and effectiveness than the identified predicate; and
The new device meets all the FDA-identified performance criteria.
If any of the above factors are not met, the submitter has the option to submit a Traditional, Special or Abbreviated 510(k).
The FDA will issue future final guidance(s) to apply this Safety and Performance Based Pathway to certain types of devices with corresponding FDA-identified performance criteria. Industry and other stakeholders may suggest device types for which the FDA should consider identifying performance criteria. For example, industry may suggest devices for which there are comprehensive FDA-recognized consensus standards. We encourage industry and other stakeholders to submit evidence-based suggestions on what the performance criteria should be for eligible device types. Input can be provided using the docket number FDA-2018-D-1387 at http://www.regulations.gov.
The FDA intends to maintain a list of device types appropriate for the Safety and Performance Based Pathway on this website, accompanied by the guidance documents that identify the performance criteria for each device type, as well as the testing methods recommended in the guidances where feasible, and any other relevant information.
Content of a Safety and Performance Based 510(k)
The amount and type of information necessary to support a finding of substantial equivalence under the Safety and Performance Based Pathway will depend on the underlying source for the performance criteria and testing methods. Table 1 and the Appendix within the Safety and Performance guidance summarize the types of information that should be included in a submission based on the submitter’s approach.
Importantly, 510(k) submitters still need to identify a predicate for certain aspects of substantial equivalence. However, instead of conducting direct comparison testing to demonstrate that a device is as safe and effective as a predicate device, manufacturers will have the option to use this pathway to demonstrate substantial equivalence, when appropriate.
More information on content that should be included within a Safety and Performance Based 510(k) will be available in the device-specific guidances that will be issued.
Thank you for visiting FWQRC blogs
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
Loading…Leave a comment
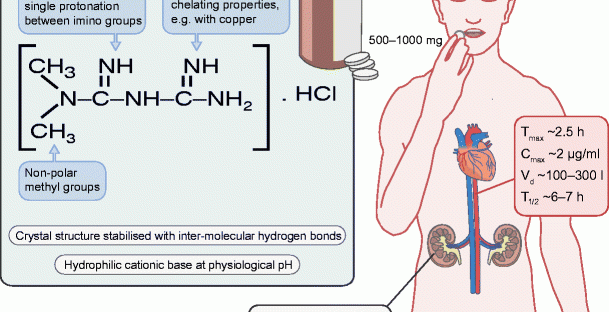
HEALTHCARE INSTITUTIONS, IMPURITIES, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
Impurities found in diabetes drugs outside the U.S
Hi, Welcome to Regulatory Focus News Letter
Here we are going to discuss about the Statement from Janet Woodcock, M.D., director of FDA’s Center for Drug Evaluation and Research, on impurities found in diabetes drugs outside the U.S
The U.S. Food and Drug Administration has been investigating the presence of genotoxic impurities, called nitrosamines, in some types of drugs. Over the past year and a half, several drug products including angiotensin II receptor blockers (ARBs) and ranitidine, commonly known as Zantac, have been found to contain small amounts of nitrosamines such as N-Nitrosodimethylamine (NDMA). During this time, there has been an ongoing investigation into the presence of nitrosamines in other drug products. This effort is focused on ensuring the drugs used by Americans continue to meet strict quality standards.
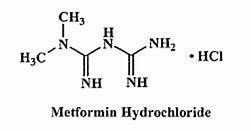
The FDA is aware that some metformin diabetes medicines in other countries were reported to have low levels of NDMA. Based on the information we have available, the levels of NDMA seen outside the U.S. are within the range that is naturally occurring in some foods and in water. While we are aware that some regulatory agencies outside the U.S. may be recalling some metformin drugs, there are no metformin recalls affecting the U.S. market at this time. The FDA is investigating whether metformin in the U.S. market contains NDMA, and whether it is above the acceptable daily intake limit of 96 nanograms. The agency will also work with companies to test samples of metformin sold in the U.S. and will recommend recalls as appropriate if high levels of NDMA are found. If as part of our investigation, metformin drugs are recalled, the FDA will provide timely updates to patients and health care professionals.

Metformin is a prescription drug used to control high blood sugar in patients with type 2 diabetes. Patients should continue taking metformin to keep their diabetes under control. It could be dangerous for patients with this serious condition to stop taking their metformin without first talking to their health care professional. The FDA recommends prescribers continue to use metformin when clinically appropriate, as the FDA investigation is still ongoing, and there are no alternative medications that treat this condition in the same way.
NDMA is a common contaminant found in water and foods including cured and grilled meats, dairy products and vegetables. Everyone is exposed to some level of NDMA. The FDA and the international scientific community do not expect it to cause harm when ingested at low levels. The acceptable daily intake limit for NDMA in the U.S. is 96 nanograms. Genotoxic substances such as NDMA may increase the risk of cancer if people are exposed to them above acceptable levels and over long periods of time, but a person taking a drug that contains NDMA at-or-below the acceptable daily intake limit every day for 70 years is not expected to have an increased risk of cancer.
Today, we have better testing methods than ever before, and we know what to look for in products’ chemical structure and manufacturing processes that may increase the risk of forming low levels of nitrosamines. Improved technology enables us to detect even trace amounts of impurities in drug products and may be the reason why more products have been found to have low levels of NDMA. The agency has strict standards for safety, effectiveness and quality, and our staff makes every effort to help keep the U.S. drug supply as safe as possible. We also work closely with international drug regulatory agencies so that we leverage resources and testing done outside the U.S. which can help inform testing of the U.S. drug supply. As our investigations and testing continues, along with the investigations done by other drug regulatory agencies, we may find low levels of nitrosamines in additional drugs.

The FDA will continue to investigate the source of these impurities, but it is important to note that there are multiple reasons why NDMA can be present in drugs. Previously, we found the source of NDMA can be related to the drug’s manufacturing process or its chemical structure or even the conditions in which they are stored or packaged. As food and drugs are processed in the body, nitrosamines, including NDMA, can be formed. The FDA continues to test and research possible sources for the several drugs found to contain NDMA.
We are taking a systematic approach to identify medicines with nitrosamines above acceptable daily intake limits and remove them from the market. For example, yesterday we announced expanded testing requirements for ranitidine manufacturers to help give consumers confidence that the drugs on the market do not have NDMA above the acceptable daily intake limit.
Our investigations, including our current investigation of metformin, take into account the medical necessity of the drug, how many Americans may take it, and whether there may be alternative treatments available. The American public can expect that we will act quickly to address any issue as soon as we find out about it.
These investigations take time. We understand that these issues affect patients’ health and well-being in many ways, and the FDA’s goal is to provide patients and health care providers as much clarity and as many answers as possible to inform their health care decisions. The FDA will communicate any information we have scientifically confirmed to ensure the public knows as much as possible as soon as possible.
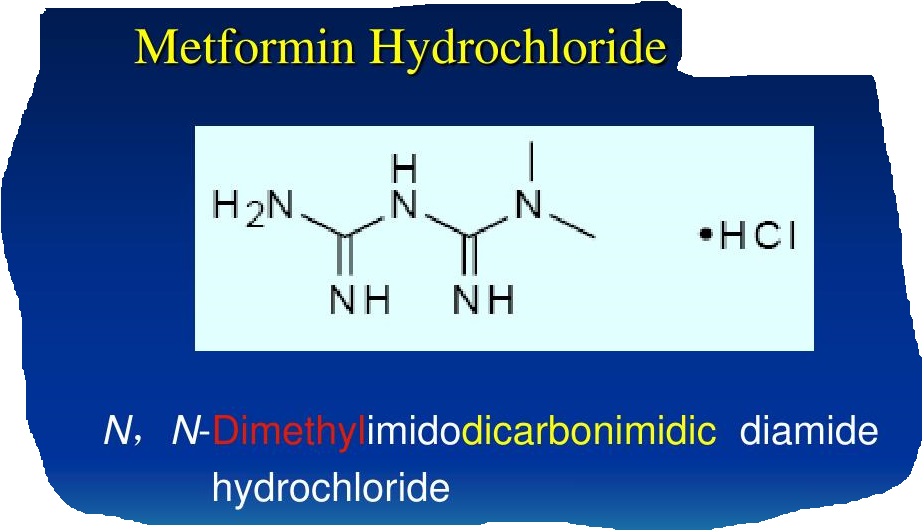
Protecting patients is the FDA’s highest priority, and Americans can be confident in the quality of the products the agency approves. We are patients too, and we’re committed to maintaining our high standards for quality, safety and efficacy for all drugs we, our families, friends, colleagues and millions of fellow Americans rely on for their health.
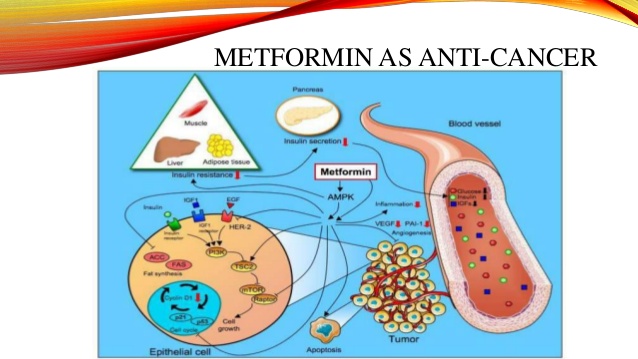
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Thank you for visiting FWQRC blogs
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
Loading…Tagged GMP ComplianceLeave a comment

HEALTHCARE INSTITUTIONS, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
FDA approves first generics of Gilenya
Hi, Welcome to Regulatory Focus News Letter from FWQRC
Here we are going to discuss about the capsules for the treatment of relapsing forms of multiple sclerosis (MS) in adult patients
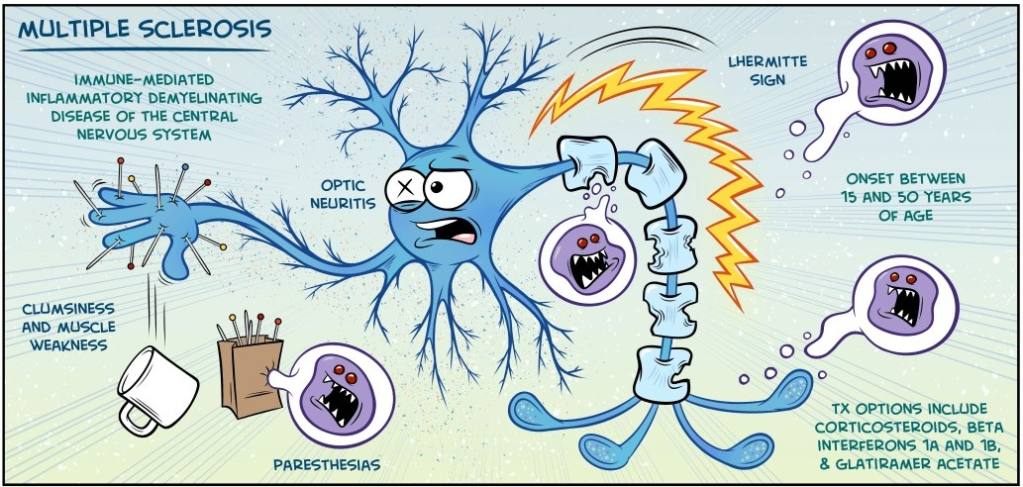
The U.S. Food and Drug Administration has approved three applications for first generics of Gilenya (fingolimod) capsules for the treatment of relapsing forms of multiple sclerosis (MS) in adult patients.
“Approving safe and effective generics so patients have more treatment options continues to be a priority for the FDA,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “Having access to affordable treatments is important for patients with conditions that require ongoing care. The FDA has a longstanding commitment to increasing patient access to lower-cost, high-quality generic medicines.”

MS is a chronic, inflammatory, autoimmune disease of the central nervous system that disrupts communication between the brain and other parts of the body. It is among the most common causes of neurological disability in young adults and occurs more frequently in women than men. For most people with MS, episodes of worsening function and appearance of new symptoms, called relapses or flare-ups, are initially followed by recovery periods (remissions). Over time, recovery may be incomplete, leading to progressive decline in function and increased disability. Gilenya is a widely used orally administered treatment option.
The most common side effects reported in the clinical trials for Gilenya include headache, elevation of liver enzymes, diarrhea, cough, influenza, sinusitis, back pain, abdominal pain and pain in the extremities.

Fingolimod must be dispensed with a Medication Guide that contains important information about its uses and risks. Serious risks include slowing of the heart rate, especially after the first dose. Fingolimod may increase the risk of serious infections. Patients should be monitored for infection during treatment and for two months after discontinuation of treatment.
A rare brain infection that usually leads to death or severe disability, called progressive multifocal leukoencephalopathy (PML) has been reported in patients being treated with the drug. PML cases usually occur in patients with weakened immune systems. Fingolimod can cause vision problems. It may increase the risk for swelling and narrowing of the blood vessels in the brain (posterior reversible encephalopathy syndrome). Other serious risks include respiratory problems, liver injury, increased blood pressure and skin cancer. Fingolimod may cause harm to a developing fetus; health care professionals should advise women of child-bearing age of the potential risk to the fetus and to use effective contraception.

The FDA granted approvals of generic fingolimod applications to HEC Pharm Co. Limited, Biocon Limited and Sun Pharmaceutical Industries Limited.
Thank you for visiting FWQRC blogs
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
Loading…Tagged GMP ComplianceLeave a comment
HEALTHCARE INSTITUTIONS, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
FDA authorizes marketing of diagnostic test that uses novel technology to detect MRSA bacteria
Hi, Welcome to Regulatory focus news letter from FWQRC
The topic which we are going to discuss below is about MRSA (Methicillin-resistant Staphylococcus aureus)
On 5th Dec 2019, the U.S. Food and Drug Administration authorized marketing of a new diagnostic test based on bacterial viability and novel technology to detect Methicillin-resistant Staphylococcus aureus (MRSA) bacterial colonization, a widespread cause of hospital-acquired infections. The cobas vivoDx MRSA diagnostic test may allow health care professionals to evaluate patients for colonization with MRSA bacteria more quickly than traditional culture-based techniques when such testing is needed
“Diagnostics that are able to provide accurate results more quickly can offer health care providers an advantage when trying to prevent and contain the spread of resistant bacteria,” said Tim Stenzel, M.D., Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “Today’s authorization adds a new tool in the fight to prevent and control MRSA in high-risk settings. The FDA remains committed to supporting efforts to address antimicrobial resistance in order to better protect patients against this ongoing public health challenge.”

MRSA is a type of bacteria that can lead to serious illness, and even death, if a patient develops an infection. According to the Centers for Disease Control and Prevention (CDC), approximately 5% of U.S. hospital patients carry the MRSA bacteria, although many of those that carry the bacteria do not develop infections. MRSA has been defined as a serious antimicrobial resistant threat by the CDC. It is resistant to many common antibiotics, which means that if infections develop they can be very challenging to treat and control. The use of active screening to detect MRSA colonization and enable implementation of infection control measures has played an important role in reducing the rates of MRSA infection. The CDC estimates that there were more than 323,000 MRSA cases in hospitalized patients in the U.S. and more than 10,000 deaths in 2017.
The cobas vivoDx MRSA test uses a new bacteriophage technology based on bioluminescence to detect MRSA from nasal swab samples in as little as 5 hours compared to 24-48 hours for conventional culture. Diagnostic tests that can more quickly and easily detect MRSA could benefit patient care and may help healthcare providers prevent the spread of MRSA. The FDA reviewed data from performance studies in which the cobas vivoDx MRSA test correctly identified MRSA in approximately 90% of samples where MRSA was present and correctly identified no MRSA in 98.6% of samples that did not have MRSA present. The cobas vivoDx MRSA test authorized today is intended to aid in the prevention and control of MRSA infections in healthcare settings and can be used to identify patients needing enhanced precautions for infection control such as isolation and additional decolonization efforts.
The FDA reviewed the cobas vivoDx MRSA test through the de novo premarket review pathway, a regulatory pathway for low-to-moderate-risk devices of a new type. Along with this authorization, the FDA is establishing special controls for tests of this type, including requirements relating to labeling and design verification and validation to address certain risks, such as false positives. When met, the special controls, along with general controls, provide a reasonable assurance of safety and effectiveness for tests of this type. This action also creates a new regulatory classification, which means that subsequent devices of the same type with the same intended use may go through the FDA’s 510(k) pathway, whereby devices can obtain clearance by demonstrating substantial equivalence to a predicate device.
The FDA granted marketing authorization of the cobas vivoDx MRSA test to Roche Molecular Systems Inc.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Thank you for following FWQRC news letters
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=626&origin=fwqrc.wordpress.com&obj_id=151501655-626-653e420f40234Tagged PHARMACEUTICAL MANUFACTUERESLeave a comment

HEALTHCARE INSTITUTIONS, LIFE SCIENCES, MEDICAL DEVICES
FDA launches app for health care professionals to report novel uses of existing medicines for patients with difficult-to-treat infectious diseases
Welcome to Regulatory focus newsletter from FWQRC
Today’s review is about the recently launched application
The U.S. Food and Drug Administration today announced the global launch of CURE ID, an internet-based repository that will allow the clinical community to report their experiences treating difficult-to-treat infectious diseases with novel uses of existing FDA-approved drugs through a website, a smartphone or other mobile device. The platform enables the crowdsourcing of medical information from health care providers to guide potentially life-saving interventions and facilitate the development of new drugs for neglected diseases. The repository is a collaboration between the FDA and the National Center for Advancing Translational Sciences (NCATS), which is part of the National Institutes of Health (NIH).
“The CURE ID application focuses on drugs for infectious diseases lacking adequate treatments, including neglected tropical diseases, emerging infectious threats and infections caused by antimicrobial-resistant organisms. When health care professionals directly input their clinical cases into the app, CURE ID allows these real-world experiences to be organized and analyzed much faster, making it easier to spot promising new uses for existing drugs,” said Amy Abernethy, M.D., Ph.D., FDA Principal Deputy Commissioner. “Our hope is that this app will serve as a connector among major treatment centers, academics, private practitioners, government facilities and other health care professionals from around the world and ultimately get treatments to patients faster.”
The repository captures clinical outcomes when drugs are used for new indications, in new populations, in new doses or in new combinations. Health care professionals generally may choose to prescribe or use a legally marketed human drug or medical device for an unapproved or uncleared use when they judge that the unapproved use is medically appropriate for an individual patient. The systematic collection of real-world experience in the app will help identify drug candidates for additional study, encourage further drug development, and may serve as a resource for practitioners making individual patient treatment decisions in the absence of established safe and effective options. Repurposing approved drugs for new clinical indications can potentially offer an efficient drug-development pathway for treatments of diseases and conditions that have few or no therapeutic options.
“The potential importance of new therapeutic opportunities from repurposing drugs can’t be understated,” said NCATS Director Christopher P. Austin, M.D. “The CURE ID platform exemplifies how collaborative efforts can spark innovations that benefit patients. This new platform harnesses the power of crowdsourcing to help gather medical observations in the field and help identify potentially effective treatments for diseases.”
The app works by collecting a simple case report form from caregivers about their experience using an approved product for an unapproved use. Health care professionals can browse from a collection of cases that have already been documented, including successful and unsuccessful treatments, in addition to viewing relevant clinical trials and those open to enrollment at clinicaltrials.gov. App users can also participate in a treatment discussion forum where they can engage with fellow providers globally. The FDA plans to reach out to health care providers in various disciplines, including infectious and tropical diseases, to encourage them to use the app.
The full launch of the app follows the release of several pilot versions after an initial innovation award was received from the U.S. Department of Health & Human Services IDEA Lab in April 2015. During the pilots, extensive user-testing was conducted in India (2015 and 2017) and South Africa (2016). Additional feedback was also collected from users in the U.S., Europe and Peru. The updated app that launched today includes the addition of a newsfeed, an improved search feature with data from 325 different infectious diseases and syndromes to choose from, and the inclusion of nearly 1,500 initial cases from clinicians and the published literature and over 18,000 clinical trials.
To download and use the CURE ID application, visit https://cure.ncats.io or download “CURE ID” from the App or Play Store.
Thanks for visiting FWQRC.
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
Loading…Leave a comment

Recent scientific and biomedical advances
Recent scientific and biomedical advances—from genomic sequencing to development of cell and gene therapies and nanotechnologies— have brought the promise of significant improvements to the health of many millions of Americans.
To date, however, we have seen little of this promise in our day-to-day lives because a large and persistent gap separates important scientific advances and the technologies needed to translate those advances into new therapies for patients and new ways to protect the public health.

The FDA’s Technology Modernization Action Plan (TMAP), described in this document, is an important step FDA is taking to address and close this gap. It describes important near-term actions that FDA is taking to modernize use of technology—computer hardware, software, data, and analytics—to advance FDA’s public health mission.
TMAP has three elements:
- modernization of FDA’s technical infrastructure;
- enhancing FDA’s capabilities to develop technology products to support its regulatory mission; and
- communication and collaboration with stakeholders to drive technological progress that is interoperable across the system and delivers value to consumers and patients.
- The TMAP provides a sturdy technological foundation for development of FDA’s ongoing strategy around data itself—a strategy for the stewardship, security, quality control, analysis, and real-time use of data—that will accelerate the path to better therapeutic and diagnostic options for patients and clinical care providers, and better tools to enhance and promote public health.
Share this:
Loading…1 Comment

COSMETIC REGULATORY COMPLIANCE
Voluntary Cosmetic Registration Program
FDA’s Voluntary Cosmetic Registration Program (VCRP) is a reporting system for use by manufacturers, packers, and distributors of cosmetic products that are in commercial distribution in the United States

About VRCP
There are two parts to the VCRP filing, described in detail in the sections below. You may participate in both parts of the program or only one part. The VCRP regulations can be found in 21 CFR, parts 710 and 720.
The VCRP applies only to cosmetic products being sold to consumers in the United States. It does not apply to cosmetic products for professional use only, such as products used in beauty salons, spas, or skin care clinics. It also does not apply to products that are not for sale, such as hotel samples, free gifts, or cosmetic products you make in your home to give to your friends.
Benefits of VCRP Participation
The VCRP assists FDA in carrying out its responsibility to regulate cosmetics marketed in the United States. Because product filings and establishment registrations are not mandatory, voluntary submissions provide FDA with the best estimate of information available about cosmetic products and ingredients, their frequency of use, and businesses engaged in their manufacture and distribution (Federal Register, vol. 73, p. 76360, and vol. 69, p. 9339).
Information from the VCRP database also has been used by the Cosmetic Ingredient Review (CIR), an independent, industry-funded panel of scientific experts, to assist the CIR Expert Panel in establishing their priorities for assessing ingredient safety as part of their ingredient safety review (Federal Register, vol. 73, p. 76360).
Some Important Things to Know

The VCRP is a voluntary registration system for cosmetic products as defined by the Federal Food, Drug, and Cosmetic Act (FD&C Act), section 201(i). Drugs are subject to different FDA registration and marketing requirements (FD&C Act, sec. 510; 21 CFR 207). Depending on the claims made, some cosmetic products may also be drugs. If a cosmetic product is also a drug, it must comply with the requirements for both cosmetics and drugs. Additional information on these types of products is available elsewhere on FDA’s website. For example, you may wish to refer to “Is It a Cosmetic, a Drug, or Both? (Or Is It Soap?).” If your products are drugs, or both cosmetics and drugs, see “Drug Registration and Listing System (DRLS & eDRLS)” and “Electronic Drug Registration and Listing Instructions.”
The VCRP is not a cosmetic approval program or a promotional tool. Cosmetics are not subject to FDA premarket approval. It is the firm’s responsibility to ensure that its cosmetic products and ingredients are safe and properly labeled, in full compliance with the law. Registration of a cosmetic establishment, assignment of an establishment registration number, filing a cosmetic product, or assignment of a CPIS number does not mean that FDA has approved the firm or its products (21 CFR 710.8 and 720.9) or that a product is a cosmetic as defined in the FD&C Act. Any representation in labeling or advertising that creates an impression of official approval because of registration or possession of a registration number is considered misleading (21 CFR 710.8 and 720.9). Misleading labeling makes a cosmetic misbranded (FD&C Act, 602(a)).
The VCRP is not part of a prior notice system for imported cosmetics. Firms importing products considered to be solely cosmetics in the United States are not required to register with FDA.
Certain information from the VCRP database is available through the Freedom of Information Act (FOIA). For example, FDA sometimes receives such requests from consumers or healthcare providers who wish to identify products that do or do not contain certain ingredients. Proprietary business information, however, is not releasable under FOIA. Please note, in late 2018, certain registration information from the VCRP, that is currently available to the public through FOIA, will be posted on the VCRP Registration Reports webpage.
The regulations authorizing this program are found in 21 CFR, parts 710 and 720.
How to Participate
1) Registering cosmetic manufacturing and/or packaging establishments. Cosmetic establishments are facilities where cosmetics are manufactured and/or packaged, not locations that house only business operations. Only owners or operators of cosmetic manufacturing or packing facilities can register their establishments, using a separate Form FDA 2511 for each facility location. Distributors cannot register an establishment (21 CFR 710.1). Domestic firms that have begun operations can register their establishments before or after their products are entered into commercial distribution and for sale to U.S. consumers. Foreign firms may voluntarily register their establishments after their products are exported for sale in the U.S. FDA assigns a registration number to each establishment location.
2) Filing Cosmetic Product Ingredient Statements (CPIS). A cosmetic manufacturer, packer, or distributor can file a statement for each product the firm has entered into commercial distribution in the United States. Use a separate Form FDA 2512 for each formulation. (If you are using printed forms, you will need both Form FDA 2512 and 2512a.) FDA assigns a CPIS number to each formulation filed in the VCRP.
3) Amending or Discontinuing a Product Formulation. A CPIS can be amended or discontinued by filing Form FDA 2512 and continuation Form FDA 2512a. Changes to a brand name or ingredients should be submitted within 60 days after the product enters commercial distribution. A CPIS should be discontinued within 180 days after discontinuance of commercial distribution becomes known to you.
Share this:
Loading…1 Comment

HEALTHCARE INSTITUTIONS, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
FDA approves first treatment for inherited rare disease
U.S. Food and Drug Administration granted approval to Givlaari (givosiran) for the treatment of adult patients with acute hepatic porphyria, a genetic disorder resulting in the buildup of toxic porphyrin molecules which are formed during the production of heme (which helps bind oxygen in the blood).

“This buildup can cause acute attacks, known as porphyria attacks, which can lead to severe pain and paralysis, respiratory failure, seizures and mental status changes. These attacks occur suddenly and can produce permanent neurological damage and death,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research. “Prior to today’s approval, treatment options have only provided partial relief from the intense unremitting pain that characterizes these attacks. The drug approved today can treat this disease by helping to reduce the number of attacks that disrupt the lives of patients.”

The approval of Givlaari was based on the results of a clinical trial of 94 patients with acute hepatic porphyria. Patients received a placebo or Givlaari. Givlaari’s performance was measured by the rate of porphyria attacks that required hospitalizations, urgent health care visits or intravenous infusion of hemin at home. Patients who received Givlaari experienced 70% fewer porphyria attacks compared to patients receiving a placebo.
Common side effects for patients taking Givlaari were nausea and injection site reactions. Health care professionals are advised to monitor patients for anaphylactic (allergic) reaction and renal (kidney) function. Patients should have their liver function tested before and periodically during treatment.
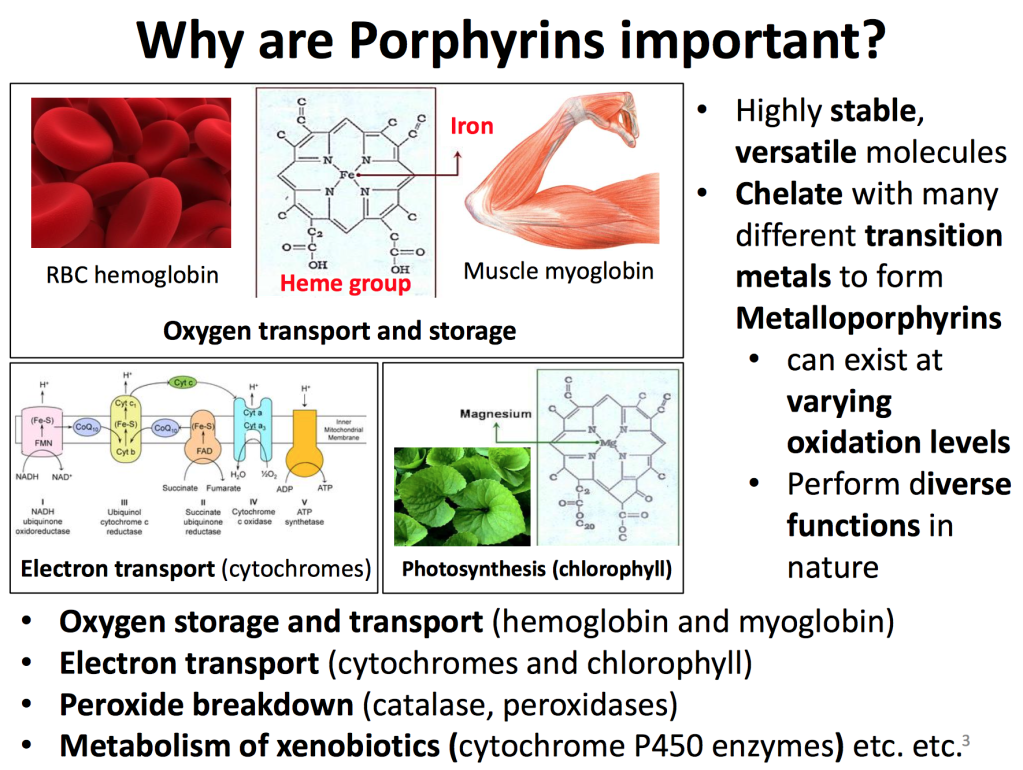
Share this:
Loading…Leave a comment
HEALTHCARE INSTITUTIONS, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
Outbreak Investigation of Hepatitis A Potentially Linked to Fresh Conventional Blackberries from Fresh Thyme Farmers Market, Fall 2019
The U.S. Food and Drug Administration (FDA), along with the Centers for Disease Control and Prevention (CDC), and state and local partners, are investigating a multistate outbreak of hepatitis A illnesses in Indiana, Nebraska, and Wisconsin potentially linked to fresh conventional (non-organic) blackberries from the grocery store, Fresh Thyme Farmers Market.
Based on the epidemiological information collected in the investigation thus far, ill patients reported consuming fresh conventional blackberries from Fresh Thyme Farmers Market stores in three states: Indiana, Nebraska, and Wisconsin.
However, traceback information to date shows that these berries came from a distribution center that ships fresh berries to Fresh Thyme Farmers Market stores in 11 states: IA, IL, IN, KY, MI, MO, MN, NE, OH, PA, and WI. As this investigation continues, the FDA will work with our federal and state partners to obtain additional information during the traceback investigation and will update this advisory as more information becomes available
Recommendation

The FDA is urging consumers to not eat any fresh conventional blackberries if purchased between September 9 and September 30, 2019, from Fresh Thyme Farmers Market stores in the 11 states mentioned above. People who purchased the fresh blackberries and then froze those berries for later consumption should not eat these berries. They should be thrown away.
If consumers purchased fresh conventional blackberries from Fresh Thyme Farmers Market stores in the 11 states listed above between September 9-30, ate those berries in the last two weeks, and have not been vaccinated for the hepatitis A virus (HAV), they should consult with their healthcare professional to determine whether post exposure prophylaxis (PEP) is indicated. PEP is recommended for unvaccinated people who have been exposed to HAV in the last two weeks. Those with evidence of previous hepatitis A vaccination or previous hepatitis A infection do not require PEP.
Contact your healthcare provider if you think you may have become ill from eating these blackberries, or if you believe that you have eaten these berries in the last two weeks
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=582&origin=fwqrc.wordpress.com&obj_id=151501655-582-653e42196a4f8Leave a comment
LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
Health Canada Creates New Medical Devices Directorate
Welcome to our Asia Regulatory Roundup, our weekly overview of the top regulatory news in Asia.
Health Canada announced the creation of a new Medical Devices Directorate to better respond to the challenges and opportunities related to the growing medical device industry.
Similar to the US FDA’s push for a lifecycle approach to regulating devices, the new Canadian devices directorate will take a lifecycle approach by bringing together postmarket functions now led by the Marketed Health Products Directorate and the pre-market functions of the Therapeutic Products Directorate.
The directorate will include 165 positions and a budget of $15.85 million funded through resource transfers and reallocation within Canada’s Health Products and Food Branch. David Boudreau, executive director at Health Canada, was named the interim director general for the new device directorate.
Health Canada said the new organization will allow for additional capacity and focus on expanding Quality Management Systems to include both internal and external components, and the implementation of ISO 9001. It will also enable better postmarket surveillance capacity for devices.
A new policy group within the directorate will help to develop new guidance tailored to medical devices, including more meaningful and targeted interactions with stakeholders, in addition to building on the expertise and activities of the newly created Digital Health Unit.
“Large-scale initiatives are underway that are challenging us to: shift how we regulate investigational trials; re-examine risk-based approaches; find ways to regulate software that allow for rapid innovation cycles; and create new pathways for innovative technologies,” Health Canada said.
Last June, Health Canada began a consultation on a set of proposed regulations intended to provide the agency with better safety information for marketed Class II, III and IV medical devices. And last December, Health Canada created a medical device action plan to improve the safety, oversight and quality of devices.
Share this:
Loading…Leave a comment

LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
China Opens Food and Drug Center of Excellence in Beijing
Welcome to our Asia Regulatory Roundup, our weekly overview of the top regulatory news in Asia
China has opened a food and drug center of excellence in Beijing. The United Nations Industrial Development Organization (UNIDO) is supporting the initiative to help China establish a harmonized, competency-based food and drug safety training system.
Representatives of UNIDO, the China International Center for Economic and Technical Exchanges and China Food and Drug Administration’s Institute of Executive Development (CFDAIED) signed off on the creation of the center of excellence earlier this year. The initiative has a total budget of $500,000, a little more than one-third of which is due to be spent this year. UNIDO reports $32,000 has been spent so far.
The investment is intended to equip China to create a country-wide food and drug safety training system that is in line with, and recognized by, international standards and organizations. Specific tasks include international training through profiling, benchmarking and capacity building.
UNIDO will help out with the overall capacity development framework and provide support with the food safety side of the project. CFDAIED will leverage the food sector work to design drug-related content, possibly with the support of UNIDO. The project is due to run until October 2021
Share this:
Loading…Leave a comment

GUIDELINES, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
Malaysia’s MDA Seeks Feedback on Orphaned Medical Device Guidance
Welcome to our Asia Regulatory Roundup, our weekly overview of the top regulatory news in Asia
Malaysia’s Medical Device Authority (MDA) has published draft guidance on medical devices that are in use but no longer registered. Products can end up in this situation when their manufacturer or authorized representative stops operating.
Such products, which MDA calls “orphaned medical devices,” pose challenges as they are in use but no longer have a representative that is responsible for them. To ensure the ongoing safe use of these devices, MDA wants healthcare facilities that use orphaned medical devices to send it a notification covering details of the product.
The notification must identify a person who is responsible for the medical device. This person must live in Malaysia and hold a top managerial position at the organization that uses the device.
MDA’s focus on the seniority of the responsible person reflects the obligations the orphaned medical device status places on healthcare facilities. In the absence of a manufacturer or typical authorized representative, the healthcare facility is responsible for the risk of using the orphaned medical device and the monitoring of its safety and performance.

The draft guidance is open for feedback until 30 November
Share this:
Loading…Leave a comment

GMP, LIFE SCIENCES, REGULATORY FOCUS NEWS LETTER
New Zealand’s Medsafe to Ban Sale of Migraine Drug Next Year
Welcome to our Asia Regulatory Roundup, our weekly overview of the top regulatory news in Asia
The New Zealand Medicines and Medical Devices Safety Authority (Medsafe) is set to ban the sale of the Cafergot migraine drug. Medsafe decided to stop the distribution of the AFT Pharmaceuticals’ product after concluding the benefits no longer outweigh the risks.
Cafergot, which contains ergotamine tartrate and caffeine, is used in the treatment of acute attacks of migraine with or without aura in adults. The drug was approved in New Zealand in the late 1960s. Other countries also used ergots such as ergotamine tartrate in the past but authorities including the United Kingdom’s National Institute for Health and Care Excellence and the German Society of Neurology now recommend against their use.

The recommendations against the use of ergots are underpinned by the lack of well-documented efficacy evidence from prospective studies and concerns that they have worse safety and tolerability profiles than other migraine drugs, such as triptans.
Adverse events linked to Cafergot in migraine are relatively rare, with the Centre for Adverse Reactions Monitoring (CARM) identifying five reports as of the end of June. One of the reports covered a case of potential pancreatitis that led the Medicines Adverse Reactions Committee (MARC) to look into the drug.
A report presented to MARC said there were “fewer reports than predicted,” although the authors think that may be because use is relatively low and, as an old drug, people may have stopped reporting side effects. The report identified close to 1,800 patients who used Cafergot in New Zealand last year.
Healthcare professionals who provided feedback for the report said Cafergot is rarely prescribed today, although one person noted they know of a “small handful” of patients who use it safely. Knowledge of those people led that healthcare professional to say, “It would be a shame to lose [Cafergot] altogether.” Some of the other healthcare professionals expressed similar sentiments.
Despite that, MARC recommended the withdrawal of Cafergot at a meeting earlier this year. That led to a Medsafe notice last week that set 1 May as the date of the withdrawal of Cafergot from the New Zealand market. The timeline reflects MARC’s recommendation of a six-month transition to enable patients to safely move to new medicines.
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=555&origin=fwqrc.wordpress.com&obj_id=151501655-555-653e421facd44Leave a comment
Asia Regulatory Roundup: China Opens Food and Drug Center of Excellence
Welcome to our Asia Regulatory Roundup, our weekly overview of the top regulatory news in Asia.
TGA Suspends Ranitidine Medicines Ahead of Possible License Cancellations
The Therapeutic Goods Administration (TGA) has suspended 23 ranitidine medicines from the Australian Register of Therapeutic Goods (ARTG). TGA suspended the products for six months but thinks there are grounds to ultimately cancel the medicines from the ARTG altogether.
Last month, TGA presented results from tests on 34 ranitidine medicines sold by 10 companies. The tests, which covered 135 batches, suggested most ranitidine medicines available in Australia contained more than 0.3 parts per million (PPM) of N-nitrosodimethylamine (NDMA), meaning they breached the internationally agreed limit for the carcinogen. Back then, TGA said it was considering “suspending the registration for products which cannot demonstrate adequate safety and quality.”
Now, TGA has revealed its response to the high levels of NDMA. The Australian regulator has put 23 ranitidine medicines on a six-month suspension that will run from 16 December to 16 June. Apotex, Arrow Pharma and Sandoz are among the companies with products affected by the suspension.
Ranitidine products absent from the list of suspended medicines include two ARTG entries sold by Arrow Pharma under the Chemists’ Own brand. The products were two of five ARTG entries with batches TGA found to contain levels of NDMA below 0.3ppm. Batches taken from two other Arrow Pharma ARTG entries and one Sandoz entry also passed the TGA test but were still suspended.
TGA uses suspensions to protect patients while giving manufacturers the chance to address issues with their products. In the case of the ranitidine medicines, if the affected companies fail to make changes to reassure TGA of the safety of their products, they face the prospect of losing their place on the ARTG permanently.
In the section on the grounds for suspension for each of the 23 products, TGA wrote that, “It is likely there are grounds for cancelling this medicine from the ARTG … on the basis that the quality of the goods is unacceptable.” The affected manufacturers now have a little more than six months to stop that happening, although in some cases TGA extends suspensions to give companies more time to fix problems with their products
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=550&origin=fwqrc.wordpress.com&obj_id=151501655-550-653e421fafe41Leave a comment

GMP, LIFE SCIENCES, MEDICAL DEVICES
Medical Devices: Current Good Manufacturing Practice Quality System Regulation—21 CFR Part 820
FDA has submitted the following proposed collection of information to OMB for review and clearance.
The CGMP quality system (QS) regulation implementing authority provided by this statutory provision is found under part 820 (21 CFR part 820) and sets forth basic CGMP requirements governing the design, manufacture, packing, labeling, storage, installation, and servicing of all finished medical devices intended for human use.
The authority for this regulation is covered under sections 501, 502, 510, 513, 514, 515, 518, 519, 520, 522, 701, 704, 801, and 803 of the FD&C Act (21 U.S.C. 351, 352, 360, 360c, 360d, 360e, 360h, 360i, 360j, 360l, 371, 374, 381, and 383).
The CGMP/QS regulation includes requirements for purchasing and service controls, clarifies recordkeeping requirements for device failure and complaint investigations, clarifies requirements for verifying/validating production processes and process or product changes, and clarifies requirements for product acceptance activities, quality data evaluations, and corrections of nonconforming product/quality problems.
Requirements are compatible with specifications in the international standards “ISO 9001: Quality Systems Model for Quality Assurance in Design/Development, Production, Installation, and Servicing.” The CGMP/QS information collections will assist FDA inspections of manufacturers for compliance with QS requirements encompassing design, production, installation, and servicing processes.
Share this:
Loading…Leave a comment

GMP, LIFE SCIENCES, PHARMA, RULES, WARNING LETTERS
Regulatory focus news letter on USFDA Warning letters – Monthly Update (Nov 2019)
A summary of USFDA Warning letter is presented below for quick reference
- In total 27 USFDA Warning letters were issued in the month of November 2019.
- 11 out of 27 related to various Pharmaceutical industries ( CGMP/Finished Pharmaceuticals/Adulterated )across the Globe
- 15 are related to Unapproved New Drugs/Misbranded/Cannabidiol (CBD) Products
- 01 is related to unapproved new drugs/misbranded
Find below some of the warning letter summarizes significant violations of current good manufacturing practice (CGMP) regulations for finished pharmaceuticals. See 21 CFR, parts 210 and 211
MARCS-CMS 581785 — SEPTEMBER 10, 2019
1. Your firm failed to establish and follow an adequate written testing program designed to assess the stability characteristics of drug products and to use results of stability testing to determine appropriate storage conditions and expiration dates (21 CFR 211.166(a)).
2. Your firm failed to clean, maintain, and, as appropriate for the nature of the drug, sanitize and/or sterilize equipment and utensils at appropriate intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements (21 CFR 211.67(a)).
3. Your firm failed to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed (21 CFR 211.192).
Hazard Analysis and Risk-Based Preventive Controls (21 CFR 117, Subpart C)
1. Your hazard analysis did not identify a known or reasonably foreseeable hazard for each type of food manufactured, processed, packed, or held at your facility to determine whether there are any hazards requiring a preventive control, as required by 21 CFR 117.130(a)(1). Specifically, your hazard analysis does not identify the hazard of recontamination with environmental pathogens at all steps where ready-to-eat (RTE) food is stored and processed while exposed to the environment in your facility after the (b)(4) step, such as at the “(b)(4),” “(b)(4),” and “(b)(4)” steps.
2. You did not identify and implement sanitation preventive controls to provide assurances that any hazards requiring a preventive control (i.e., recontamination with environmental pathogens) will be significantly minimized or prevented and the food manufactured, processed, packed, or held by your facility will not be adulterated under section 402 of the FD&C Act, as required by 21 CFR 117.135(a)(1) and (c)(3), as evidenced by the following:
a. From April 2019 to May 2019, you found five (5) environmental samples positive for L. monocytogenes in the environment in Zone 3, including areas within your RTE production area where RTE food is exposed to the environment. In addition, FDA laboratory analysis of environmental sample #1088400 collected at your facility on May 21, 2019, revealed that one (1) environmental swab in Zone 3 was positive for L. monocytogenes
b. Your written corrective action procedure for your “Environmental Microbial Sampling SOP,” issued 3/19/2019, states that “(b)(4).” However, between April 2019 and May 2019, you identified five positive environmental samples of L. monocytogenes, and you do not have records to show that you conducted an investigation of the potential source or cause of contamination.
Find below some of the warning letters related to Unapproved drugs / Misbranded products
Dietary Supplement Labeling
Information on your website at https://koicbd.com suggests that you intend to market your “KOI CBD Infused Shot” product as a dietary supplement that contains CBD because the “KOI CBD Infused Shot” is described as a “supplement.” However, your product cannot be a dietary supplement because it does not meet the definition of a dietary supplement under section 201(ff) of the FD&C Act, 21 U.S.C. 321(ff). FDA has concluded, based on available evidence, that CBD products are excluded from the dietary supplement definition under sections 201(ff)(3)(B)(i) and (ii) of the FD&C Act, 21 U.S.C. 321(ff)(3)(B)(i) and (ii). Under those provisions, if an article (such as CBD) is an active ingredient in a drug product that has been approved under section 505 of the FD&C Act, 21 U.S.C. 355, or has been authorized for investigation as a new drug for which substantial clinical investigations have been instituted and for which the existence of such investigations has been made public, then products containing that substance are outside the definition of a dietary supplement.1 There is an exception if the substance was “marketed as” a dietary supplement or as a conventional food before the new drug investigations were authorized; however, based on available evidence, FDA has concluded that this is not the case for CBD. FDA is not aware of any evidence that would call into question its current conclusion that CBD products are excluded from the dietary substance definition under sections 201(ff)(3)(B)(i) and (ii) of the FD&C Act, but you may present FDA with any evidence bearing on this issue.
Unapproved New Drugs
Based on our review of your website, your “CBD HEALING BALM,” “CBD VAPE OIL,” “FULL SPECTRUM CBD TINCTURE,” “KOI LOTION,” “KOI CBD Gummies,” and “KOI CBD Infused Shot” products are drugs under section 201(g)(1) of the FD&C Act, 21 U.S.C. 321(g)(1), because they are intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease, and/or intended to affect the structure or any function of the body.
Examples of claims observed on your website, https://koicbd.com, that establish the intended use of your products as drugs include, but may not be limited to, the following:
On your webpage titled “8 Proven Benefits of CBD”:
- “CBD RELIEVES PAIN AND INFLAMMATION”
- “studies show that CBD prevents human experimental psychosis and is effective in open case reports and clinical trials in patients with schizophrenia, with a remarkable safety profile.”
- “Not only does the research show that CBD benefits including being effective in fighting breast cancer cells, data also suggest that it can be used to inhibit the invasion of lung and colon cancer, plus it possesses anti-tumor properties in gliomas and has been used to treat leukemia.”
- “CBD LOWERS INCIDENCE OF DIABETES”
On your webpage titled “IS CBD RIGHT FOR YOU?”:
- “several pre-clinical reports showing anti-tumor effects of CBD…have found reduced [cancer] [sic] cell viability, increased cancer cell death, decreased tumor growth, and inhibition of metastasis.”
On your webpage titled “CBD AND OPIOID ADDICTION”:
- “CBD FOR OPIOID ADDICTION”
- “A potential new treatment for opioid addiction has been found in a new review of previous research of cannabidiol (CBD).”
On your webpage titled “10 LITTLE KNOWN USES FOR CBD”:
- “PTSD”
- “Fibromyalgia”
- “Schizophrenia”
- “Diabetes”
- “MS”
- “Crohn’s Disease”
- “Opioid Addiction”
- “The advantage of cannabidiol as a potential treatment for opioid addiction is that it doesn’t give users a high and thus doesn’t involve a risk of misuse.”
Your “CBD HEALING BALM,” “CBD VAPE OIL,” “FULL SPECTRUM CBD TINCTURE,” “KOI LOTION,” “KOI CBD Gummies,” and “KOI CBD Infused Shot” products are not generally recognized as safe and effective for the above referenced uses and, therefore, the products are “new drugs” under section 201(p) of the FD&C Act, 21 U.S.C. 321(p). New drugs may not be legally introduced or delivered for introduction into interstate commerce without prior approval from the FDA, as described in sections 301(d) and 505(a) of the FD&C Act, 21 U.S.C. 331(d) and 355(a). FDA approves a new drug on the basis of scientific data and information demonstrating that the drug is safe and effective. There are no FDA-approved applications in effect for any of the above mentioned products.
Misbranded Drugs
Your “CBD HEALING BALM,” “CBD VAPE OIL,” “FULL SPECTRUM CBD TINCTURE,” “KOI LOTION,” “KOI CBD Gummies,” and “KOI CBD Infused Shot” products are also misbranded within the meaning of section 502(f)(1) of the FD&C Act, 21 U.S.C. 352(f)(1), in that their labeling fails to bear adequate directions for use. “Adequate directions for use” means directions under which a layperson can use a drug safely and for the purposes for which it is intended. (See 21 CFR 201.5.) The aforementioned products are offered for conditions that are not amenable to self-diagnosis and treatment by individuals who are not medical practitioners; therefore, adequate directions for use cannot be written so that a layperson can use these drugs safely for their intended purposes. FDA-approved prescription drugs that bear their FDA-approved labeling are exempt from the requirements that they bear adequate directions for use by a layperson. However, your products are not exempt from the requirement that their labeling bear adequate directions for use, 21 CFR 201.100(c)(2) and 201.115, because no FDA-approved applications are in effect for them. The introduction or delivery for introduction into interstate commerce of these misbranded drugs violates section 301(a) of the FD&C Act, 21 U.S.C. 331(a).
Prohibited Act under 301(ll) and Adulterated Human Foods
We note that your “KOI CBD Gummies” product appears to be promoted as a conventional human food. Specifically, your website refers to the gummies as “delicious, edible CBD snacks.” However, you should be aware that it is a prohibited act under section 301(ll) of the FD&C Act, 21 U.S.C. 331(ll), to introduce or deliver for introduction into interstate commerce any food to which has been added a drug approved under section 505 of the FD&C Act or for which substantial clinical investigations have been instituted and for which the existence of such investigations has been made public. Based on available evidence, FDA has concluded that the prohibition in section 301(ll) applies to CBD. There is an exception if the substance was marketed in food before the drug was approved or before the substantial clinical investigations involving the drug had been instituted. However, based on the available evidence discussed above, FDA has concluded that this is not the case for CBD. FDA is not aware of any evidence that would call into question its current conclusion that section 301(ll) of the FD&C Act prohibits the introduction into interstate commerce of any food to which CBD has been added, but you may present FDA with any evidence bearing on this issue.
You should also be aware that, as defined in section 201(s) of the FD&C Act (21 U.S.C. 321(s)), the term “food additive” refers to any substance the intended use of which results in its becoming a component of any food, unless the substance is generally recognized as safe (GRAS) among qualified experts under the conditions of its intended use, or unless the substance meets a listed exception.2
Food additives require premarket approval based on data demonstrating safety. Any food additive that has not been approved for its intended use in food is deemed to be unsafe under section 409(a) of the FD&C Act (21 U.S.C. 348(a)), and causes the food to be adulterated under section 402(a)(2)(C)(i) of the FD&C Act, 21 U.S.C. 342(a)(2)(C)(i). Introduction of an adulterated food into interstate commerce is prohibited under section 301(a) of the FD&C Act, 21 U.S.C. 331(a).
There is no food additive regulation which authorizes the use of CBD. We are not aware of any information to indicate that CBD is the subject of a prior sanction (see 21 CFR Part 181). Furthermore, we are not aware of any basis to conclude that CBD is GRAS for use in conventional foods. FDA’s regulations in 21 CFR 170.30(a)-(c) describe the criteria for eligibility for classification of a food ingredient as GRAS. The use of a food substance may be GRAS based on either scientific procedures or, for a substance used in food before 1958, through experience based on common use in food (see 21 CFR 170.30).
We know of no basis for general recognition of safety for CBD based either on scientific procedures or common use in food prior to January 1, 1958. Based on our review of published, scientific literature, existing data and information do not provide an adequate basis to conclude that the use of CBD in food meets the criteria for GRAS status. Many unanswered questions and data gaps about CBD toxicity exist, and some of the available data raise serious concerns about potential harm from CBD. Our review of publicly available data associated with the one FDA-approved CBD drug, as well as our review of published scientific literature, identified potential for liver injury from CBD and potentially harmful interactions with certain drugs. In addition, studies in animals have shown that CBD can interfere with the development and function of testes and sperm, decrease testosterone levels, and impair sexual behavior in males. Therefore, based on our review, the use of CBD in conventional food products does not satisfy the criteria for GRAS status under 21 CFR 170.30.
FDA is not aware of any other exception to the food additive definition that would apply to CBD for use as an ingredient in a conventional food. Therefore, CBD added to a conventional food is a food additive under section 201(s) of the FD&C Act and is subject to the provisions of section 409 of the FD&C Act. Under section 409, a food additive is deemed unsafe unless it is approved by FDA for its intended use prior to marketing. CBD is not approved for use in any conventional food. Food containing an unsafe food additive within the meaning of section 409 is adulterated within the meaning of section 402(a)(2)(C)(i). Therefore, to the extent that you intend to market this products as a food, your “KOI CBD Gummies” product is also adulterated within the meaning of section 402(a)(2)(C)(i) of the FD&C Act. Introduction of an adulterated food into interstate commerce is prohibited under section 301(a) of the FD&C Act, 21 U.S.C. 331(a).
Unapproved New Animal Drugs
During our review of your website, https://koicbd.com, FDA determined that your firm is marketing the unapproved new animal drugs “KOI Naturals CBD Spray for Pets” and “KOI CBD Soft Chews.” Based on our review of your website, your “KOI Naturals CBD Spray for Pets” and “KOI CBD Soft Chews” products are drugs under section 201(g)(1) of the FD&C Act, 21 U.S.C. 321(g)(1), because they are intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in animals and/or intended to affect the structure or any function of the body of an animal. Further, as discussed below, these products are unapproved new animal drugs and marketing them violates the FD&C Act.
Examples of claims observed on your firm’s website https://koicbd.com that show the intended uses of these products include, but are not limited to, the following:
On your webpage titled “Is CBD Oil Good for Pets”:
- “Do you have a pet who is exhibiting notable discomfort? Consider giving them CBD oil to alleviate their symptoms.”
- “So although it helps to alleviate pain, there is no risk of addiction or psychoactive side effects.”
These products are “new animal drugs” under section 201(v) of the FD&C Act, 21 U.S.C. 321(v), because they are not generally recognized, among experts qualified by scientific training and experience to evaluate the safety and effectiveness of animal drugs, as safe
and effective for use under the conditions prescribed, recommended, or suggested in the labeling.
To be legally marketed, a new animal drug must have an approved new animal drug application, conditionally approved new animal drug application, or index listing under sections 512, 571, and 572 of the FD&C Act, 21 U.S.C. 360b, 360ccc, and 360ccc-l. These products are not approved or index listed by the FDA, and therefore these products are considered unsafe under section 512(a) of the FD&C Act, 21 U.S.C. 360b(a), and adulterated under section 501(a)(5) of the FD&C Act, 21 U.S.C. 351(a)(5). Introduction of these adulterated drugs into interstate commerce is prohibited under section 301(a) of the FD&C Act, 21 U.S.C. 331(a).
301(ll) and Adulterated Animal Foods
Moreover, to the extent that you market any of your products containing CBD as animal food, you should be aware that it is a prohibited act under section 301(ll) of the FD&C Act, 21 U.S.C. 331(ll), to introduce or deliver for introduction into interstate commerce any animal food to which has been added a drug approved under section 505 of the FD&C Act or for which substantial clinical investigations have been instituted and for which the existence of such investigations has been made public. Based on available evidence, FDA has concluded that the prohibition in section 301(ll) applies to CBD, as described above.
You should also be aware that, as defined in section 201(s) of the FD&C Act (21 U.S.C. 321(s)), the term “food additive” refers to any substance the intended use of which results in its becoming a component of any animal food, unless the substance is generally recognized as safe (GRAS) among qualified experts under the conditions of its intended use, or unless the substance meets a listed exception.3
There is no animal food additive regulation that authorizes the use of CBD. We are not aware of any information to indicate that CBD is the subject of a prior sanction (i.e., a sanction or approval granted prior to the enactment of the Food Additives Amendment of 1958 under the FD&C Act, the Poultry Products Inspection Act, or the Meat Inspection Act). Furthermore, we are not aware of any basis to conclude that CBD is GRAS for use in animal foods. FDA’s regulations in 21 CFR 570.30(a)-(c) describe the criteria for eligibility for classification of an animal food ingredient as GRAS. The use of an animal food substance may be GRAS based on either scientific procedures or, for a substance used in animal food before 1958, through experience based on common use in animal food (see 21 CFR 570.30). We know of no basis for general recognition of safety for CBD based either on scientific procedures or common use in animal food prior to January 1, 1958. Based on our review of the publicly available literature, the data and information necessary to support the safe use of CBD in animal foods are lacking. In fact, literature reports have raised safety concerns for animals consuming CBD, including, but not limited to, male reproductive toxicity and liver toxicity. Therefore, based on our review, the use of CBD in animal products does not satisfy the criteria for GRAS status under 21 CFR 570.30.
Under section 409 of the FD&C Act, 21 U.S.C. 348, an animal food additive is deemed unsafe unless it is approved by FDA for its intended use prior to marketing. CBD is not approved for use in any animal food. Animal food containing an unsafe food additive within the meaning of section 409 is adulterated within the meaning of section 402(a)(2)(C)(i) of the FD&C Act. Introduction of an adulterated animal food into interstate commerce is prohibited under section 301(a) of the FD&C Act, 21 U.S.C. 331(a).
Share this:
Loading…Leave a comment

GMP, GUIDELINES, LIFE SCIENCES
CVM GFI #256 – Compounding Animal Drugs from Bulk Drug Substances
On 19th Nov 2019 FDA circulated draft guideline for compounding animal drugs from Bulk drug Substances for public review comments. The comments shall be submitted to FDA through online by 18th Feb 2020
This draft guidance is intended for veterinarians, State-licensed pharmacies, and Federal facilities interested in compounding animal drugs from bulk drug substances for use in nonfood-producing animals or for use as antidotes in food-producing animals under limited circumstances when no other medically appropriate treatment options exist.
Under the Federal Food, Drug, and Cosmetic Act (FD&C Act), the compounding of an animal drug from bulk drug substances results in a “new animal drug” that must comply with the FD&C Act’s animal drug approval, conditional approval, or indexing requirements (sections 512, 517, and 572 of the FD&C Act). In addition, all animal drugs are require to, among other things, be made in accordance with current good manufacturing practice (cGMP) requirements (section 501(a)(2)(B) of the FD&C Act) and have adequate directions for use (section 502(f)(1) of the FD&C Act).
Animal drugs that are compounded from bulk drug substances do not meet the FD&C Act’s new animal drug approval, cGMP, or adequate directions for use requirements. However, FDA has generally exercised enforcement discretion with regard to animal drug compounding from bulk drug substances under certain circumstances. This draft guidance is a continuation of this practice and is intended to provide additional information and clarity to veterinarians and pharmacists about FDA’s current thinking on this matter. The guidance describes the circumstances under which FDA, at this time and based on our current understanding of the risks of animal drugs compounded from bulk drug substances, does not intend to take enforcement action for violations of the FD&C Act with respect to the compounding of animal drugs from bulk drug substances.
Contact FWQRC™ for GMP Training, Auditing by QP, eCTD, GAP Analysis, Risk Assessment, CAPA, CSV, Method development/Validation, ADE/PDE Values, Facility & Product Registrations
Share this:
Loading…1 Comment

FDA Releases 105 Product-Specific Guidances
The US Food and Drug Administration (FDA) on Thursday released 105 product-specific guidance documents to aid generic drug development, including 27 new draft guidances and 78 revised draft guidances.
The guidances, when finalized, are intended to promote generic competition by clarifying FDA’s expectations for the studies required to demonstrate that a generic drug is equivalent to a reference listed drug. So far, FDA has issued nearly 1,800 product-specific guidances.
As part of its commitments under the Generic Drug User Fee Amendments (GDUFA II) reauthorization, FDA committed to issuing product-specific guidances for 90% of non-complex new chemical entity new drug applications (NDAs) approved after 1 October 2017 at least two years ahead of the earliest abbreviated new drug application (ANDA) filing date. The agency also committed to issuing product-specific guidances for complex products “as soon as scientific recommendations are available.”
Among the new guidances are recommendations for the studies necessary to support ANDA approvals for Viiv Healthcare’s two-drug once-daily HIV drug Juluca (dolutegravir/rilpivirine), Amicus Therapeutics’ Fabry disease drug Galafold (migalastat) and Advanced Accelerator Applications’ cancer drug Lutathera (lutetium dotatate Lu-177).
Most of the new draft guidances are for oral formulations, though a few are for ophthalmic, intravenous and subcutaneous formulations.
Most of the revised draft guidances are for topical and transdermal formulations, including extended release film formulations of estradiol, fentanyl and nicotine. In an email to Focus, FDA spokesperson Charles Kohler said that most of the revisions “are minor or editorial in nature, meant to ensure the PSGs are consistent, clear, up-to-date, and include links to the most current related guidances.”
Share this:
Loading…Leave a comment

New catalyst efficiently produces hydrogen from seawater
Seawater is one of the most abundant resources on earth, offering promise both as a source of hydrogen – desirable as a source of clean energy – and of drinking water in arid climates. But even as water-splitting technologies capable of producing hydrogen from freshwater have become more effective, seawater has remained a challenge.

Researchers from the University of Houston have reported a significant breakthrough with a new oxygen evolution reaction catalyst that, combined with a hydrogen evolution reaction catalyst, achieved current densities capable of supporting industrial demands while requiring relatively low voltage to start seawater electrolysis.
Researchers say the device, composed of inexpensive non-noble metal nitrides, manages to avoid many of the obstacles that have limited earlier attempts to inexpensively produce hydrogen or safe drinking water from seawater. The work is described in Nature Communications.
Zhifeng Ren, director of the Texas Center for Superconductivity at UH and a corresponding author for the paper, said a major obstacle has been the lack of a catalyst that can effectively split seawater to produce hydrogen without also setting free ions of sodium, chlorine, calcium and other components of seawater, which once freed can settle on the catalyst and render it inactive. Chlorine ions are especially problematic, in part because chlorine requires just slightly higher voltage to free than is needed to free hydrogen.
The researchers tested the catalysts with seawater drawn from Galveston Bay off the Texas coast. Ren, M.D. Anderson Chair Professor of physics at UH, said it also would work with wastewater, providing another source of hydrogen from water that is otherwise unusable without costly treatment.
“Most people use clean freshwater to produce hydrogen by water splitting,” he said. “But the availability of clean freshwater is limited.”
To address the challenges, the researchers designed and synthesized a three-dimensional core-shell oxygen evolution reaction catalyst using transition metal-nitride, with nanoparticles made of a nickle-iron-nitride compound and nickle-molybdenum-nitride nanorods on porous nickle foam.
First author Luo Yu, a postdoctoral researcher at UH who is also affiliated with Central China Normal University, said the new oxygen evolution reaction catalyst was paired with a previously reported hydrogen evolution reaction catalyst of nickle-molybdenum-nitride nanorods.
The catalysts were integrated into a two-electrode alkaline electrolyzer, which can be powered by waste heat via a thermoelectric device or by an AA battery.
Cell voltages required to produce a current density of 100 milliamperes per square centimeter (a measure of current density, or mA cm-2) ranged from 1.564 V to 1.581 V.
The voltage is significant, Yu said, because while a voltage of at least 1.23 V is required to produce hydrogen, chlorine is produced at a voltage of 1.73 V, meaning the device had to be able to produce meaningful levels of current density with a voltage between the two levels.
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=514&origin=fwqrc.wordpress.com&obj_id=151501655-514-653e42236a5a7Leave a comment

Announcement to all CEP holders for synthesised APIs regarding presence of nitrosamines
Since July 2018, EDQM has been actively involved in activities related to the detection and control of nitrosamine impurities in sartan active substances (APIs) with a tetrazole ring for which there are CEPs. These activities have included contacting CEP holders to request data and corrective actions, assessing responses to such requests, GMP inspections of manufacturing sites, suspension of CEPs, where appropriate, and their restoration after appropriate corrective actions had been implemented. Additionally, for any APIs, for new applications for a CEP, during the renewal of a CEP or during any revisions where the synthetic route or sourcing is modified, the potential for presence of such nitrosamine impurities is systematically assessed. EDQM has also been involved in the activities of the OMCL network including coordination of sampling and testing. The Ph. Eur. monographs for the sartan APIs with a tetrazole ring have been revised as a consequence of the presence of nitrosamines in these substances.
As has been announced on the EMA and EDQM website recently, the presence of a nitrosamine impurity has recently been identified in ranitidine-containing medicinal products. As a result of this, action has been taken to suspend the relevant CEPs and more information is awaited to better understand the root cause of the presence of this nitrosamine impurity.
EMA and CMDh have published documents on their websites, (EMA/189634/2019 and CMDh/404/2019), “Information on nitrosamines for marketing authorisation holders” which request marketing authorisation holders (MAH) to follow an investigation process described for synthesised APIs (other than sartans with a tetrazole ring). Although EDQM does not expect that this issue impacts many substances, it is now appropriate to expand the review to all other APIs manufactured from chemical synthesis for which CEPs have been granted. EDQM therefore requests that the holders of such CEPs follow the process described here (which is a similar stepwise approach to that for MAH):
Step 1 – Risk evaluation: Companies holding CEPs should perform a risk evaluation of their chemically synthesised APIs with regards nitrosamine formation, using quality risk management principles, as outlined in the ICH Q9 guideline. The principles described in the ICH M7 guideline in relation to toxicology assessment, control strategy and changes to the manufacturing process for active substances should be applied.
The CEP holders should prioritise substances in order to establish the sequence in which they are to be evaluated. The factors that can be taken into account are outlined in the dedicated Questions and Answers documents EMA/CHMP/428592/2019 Rev. 1 and CMDh/405/2019, Rev.1 which are available on the EMA and CMDh websites and if any substances are identified as high priority, the risk evaluation should be done immediately. The risk evaluation should address not only risks from the manufacturing process but also those from the introduction of materials used in the manufacturing process (e.g. starting materials, reagents, solvents – fresh and recovered etc.).
The risk evaluation for all CEPs should be concluded at the latest by 26th March 2020.
CEP holders are not required to confirm to EDQM that step 1 has been completed where no risk is identified. However where a risk is identified, EDQM should be immediately informed and the company should progress to step 2, confirmatory testing, and the timescales for when the confirmatory testing results will be provided to EDQM should be indicated to EDQM.
The outcome of step 1 relating to the risk evaluation performed for a CEP should be communicated to the customers in all cases (even if no risk is identified) such that the MAHs can use this information to fulfil their responsibilities as described in the documents, EMA/189634/2019 and CMDh/404/2019 mentioned earlier.
Step 2 – Confirmatory testing: in the event that a risk of presence of nitrosamines is identified as a result of the risk evaluation, confirmatory testing should be carried out using appropriately validated and sensitive methods in accordance with the prioritisation deriving from the risk evaluation conducted in step 1. Substances identified as high priority should be tested as soon as possible.
All APIs identified to be at risk of presence of nitrosamines should be tested and the results provided to EDQM with, if needed, a proposal for subsequent actions (such as revision of the CEP).
CEP holders should inform the EDQM immediately if the tests confirm the presence of a nitrosamine impurity, irrespective of the amount detected, and provide the results. The companies should then progress to step 3 after informing EDQM of the plan and timescales to complete step 3.
Step 3 – Revision to the CEP: Where nitrosamine impurities have been detected, CEP holders should apply for a revision to their application(s) in a timely manner to introduce any required changes, such as amendment of the manufacturing process or changes to specifications and introduction of controls.
The required revisions to the CEP applications should be concluded at the latest by 26th September 2022 or at an earlier time if otherwise justified.
CEP holders are reminded that it is their responsibility to complete the procedure described in this announcement for all their impacted CEPs.
EDQM may contact CEP holders for information on the risk of nitrosamines at any step of this review and any such requests should be responded to fully and in a timely manner. EDQM will take action on any CEP (e.g. suspension) where information becomes available regarding an unacceptable level of nitrosamine impurities in the active substance which is the subject of a CEP.
EDQM reminds CEP holders that they should provide the appropriate information relating to the risk evaluation they have performed for their CEP to their customers in all cases (for steps 1 to 3, and for step 1 even if no risk has been identified) such that the MAHs can use this information to fulfil their responsibilities in a timely manner as described in the documents, EMA/189634/2019 and CMDh/404/2019 mentioned earlier.
Share this:
Loading…Leave a comment

Fall Regulatory Agenda: FDA Delays Release of Several Proposed Rulemakings
The latest regulatory agenda for the US Food and Drug Administration (FDA) was released on Wednesday, announcing the delays of several proposed rulemakings, and one new but expected addition on the importation of prescription drugs.
On importation, the proposed rule, which has been under review at the Office of Management and Budget since the beginning of November and is expected to be released by January, would allow pharmacists and wholesalers to import prescription drugs from Canada if the imports pose no additional risks and will save money.
The proposed rule is expected to have several key limitations (i.e. only facilities that also manufacture API for the FDA-approved version would be allowed) and exclusions (no costly biologics, drugs with a REMS or infused or injected drugs) that may raise questions about what drugs could be imported and how much could be saved.
And as the latest agenda says, this regulatory action “will be likely to have international trade and investment effects,” and Canadian pharmacists and Health Canada officials have pushed back against the proposal. FDA is also expected to release draft guidance to allow manufacturers to voluntarily import US versions of drugs sold in foreign countries.
Some Proposed Rulemakings Delayed
All of the other proposed and final rulemakings in the fall regulatory agenda were previously published in prior agendas, including the agenda from last May. But this latest agenda shows how a few of the proposed rulemakings are being held up.
For instance, in April 2020, FDA expects to publish a proposed rule on the modernization of the quality system regulation for medical devices. FDA previously said it would publish the proposed rule last September.
FDA also delayed its proposed rulemaking on excluding certain medical software functions from the definition of a device until next May. The agency previously said the proposed rule was coming in December.
The proposed rulemaking on the annual summary reporting requirements under the Right to Try Act were also delayed from last September to sometime this month.
The agency further held the release of its rulemaking on post approval changes to approved drug and biologic applications from next March to next September.
The fall agenda also includes a proposed rulemaking on pediatric study plan requirements for new drug and biologic applications, which is due for release in September 2020, but which had a legal deadline of July 2013.
And another proposed rulemaking to better define and clarify the roles and responsibilities of those engaged in the initiation, conduct and oversight of clinical investigations subject to IND requirements was also pushed back from April 2020 to September 2020.
Other proposed rulemakings have stayed the course since last May, including one on clinical holds in medical device investigations, which is due in February 2020, one on biologics regulation modernization, which is still slated for December and one on patent term restoration due in February 2020.
Final Rules
Some of the final rules expected to be released later this year or in 2020 also reveal how long or how quickly it can take a rulemaking to go from proposal to finalization.
For instance, a proposed rulemaking on changes to the way risks are presented in direct-to-consumer drug advertising may end up taking more than a decade to go from proposal through three comment periods to finalization next June.
A proposed rulemaking on postmarket safety reporting requirements for drugs and biologics has been lingering since 2003, but is expected to be re-proposed next June.
In contrast, the so-called “deemed to be a license” rule transitioning some new drug applications to biologic license applications is expected to go from proposal to finalization in less than a year. Similarly, a proposed rule on the de novo classification process for medical devices is expected to be finalized in June after being first proposed last December.
Fall 2019 Unified Agenda of Regulatory and Deregulatory Actions
Share this:
Loading…Leave a comment

FDA Raises Concerns With API Manufacturers
As quality issues have led to drug shortages, Donald Ashley, director of FDA’s Office of Compliance, raised several major concerns on Tuesday with the active pharmaceutical ingredient (API) industry, noting three trends related to the obfuscation of supply chain information, an increasing number of data integrity question marks and impurity concerns that have led to recalls.
On the obfuscation front, Ashley, speaking at the Association for Accessible Medicines’ conference in Bethesda, MD, noted that API companies sometimes fail to obtain and retain documents with the identity of the original manufacturer and certificate of analysis.
In addition, APIs, including opioids, are often distributed with inadequate certificates of analysis, which he said, “compromises supply chain accountability and traceability and may put consumers at risk.”
On data integrity, which often includes incomplete, inconsistent or inaccurate data, Ashley noted that of the warning letters issued to API manufacturers over the past four years, 73% have included including data integrity charges.
He also quoted from a warning letter sent to API manufacturer Lantech Pharmaceuticals in August, in which the firm admitted to “routinely deleting recovered solvents gas chromatography data older than three months permanently, without any backup.”
Ashley added: “Data integrity problems you can see are the tip of the iceberg,” noting that lapses often obscure other problems.
He also highlighted ICH’s Q7 guideline, which deals with the cGMPs for APIs, and which explains: “Agents, brokers, distributors, repackers, or relabelers should transfer all quality or regulatory information received from an API or intermediate manufacturer to the customer, and from the customer to the API or intermediate manufacturer.”
On the impurities front, he also advised companies to consult ICH guidelines Q3A, Q3B(R2), Q3C, Q3D and M7(R1), as N-Nitrosodimethylamine (NDMA) and N-Nitrosodimethylamine (NDEA) have been found in a few different medicines. Recalls of the heartburn medicine ranitidine and the blood pressure medicine valsartan have occurred because of such impurities. However, FDA recently downplayed the levels of impurities in some ranitidine medicines, noting they “are similar to the levels you would expect to be exposed to if you ate common foods like grilled or smoked meats.”
Outside the API concerns, Ashley noted that overall, the number of warning letters issued by FDA has increased between FY 2015 and 2019, although the agency has seen improvements in reducing the time between when an inspection ends and when a warning letter is issued, from a median of 11.6 months in 2015 to 6.5 months in 2019. He noted FDA is hoping to bring the time between inspection and issuance of the warning letter to six months by FY 2020.
Share this:
Loading…Leave a commentGMP
FDA issues final guidance for development of smallpox treatments as part of preparedness efforts
“While the World Health Organization declared smallpox eradicated in 1980, concerns have persisted that smallpox could be used as a biological weapon. The FDA plays a pivotal role preparing our nation to be able to protect the American people from biological threats, including by providing guidance and support for the development of medical countermeasures that can be used safely, effectively and reliably during public health emergencies,” said Anna Abram, FDA Deputy Commissioner for Policy, Legislation, and International Affairs.
“We work with government partners as well as non-government organizations, universities and research centers, and industry to further the development of medical countermeasures as part of our vital public health mission. Despite recent advances in developing an effective treatment for smallpox, drug developers still face challenges in bringing forward these medical countermeasures, which are critical should smallpox ever be used as a biological weapon. The agency’s work to advance safe and effective medical countermeasures is a high priority, and today’s final guidance on the development of drugs to treat or prevent smallpox builds upon currently available guidance, further advancing the agency’s long-standing commitment to the development of robust medical countermeasure preparedness efforts.”
On November 15, 2019, FDA issued final guidance, Smallpox (Variola Virus) Infection: Developing Drugs for Treatment or Prevention, which is designed to assist drug manufacturers designing studies to appropriately establish the safety and efficacy of drugs to treat or prevent smallpox infection.
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=495&origin=fwqrc.wordpress.com&obj_id=151501655-495-653e422a2e747Leave a commentGMP
Ebola survivor study expanded to include new technologies

In addition to supporting ongoing response to Ebola outbreaks in the Democratic Republic of the Congo (DRC), FDA and government partners are conducting studies in West Africa to better understand how Ebola affects patients who have survived, and to learn how to more effectively treat these patients’ chronic health problems
In 2016, FDA awarded a contract to Stanford University to help the global scientific community better understand the course of Ebola virus infection—an important factor in finding new treatments. In 2017, research was expanded to include Zika virus infection.
In September 2019, the project was again expanded, to apply a new method to the study of Ebola and Zika tissue samples. The Stanford laboratory will use multiplexed ion beam imaging (MIBI) to identify viral reservoirs—cells or anatomical sites where viruses accumulate and persist—for both Ebola and Zika infection, ultimately facilitating the deployment of novel, effective analytical technologies into federal laboratory space.
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=490&origin=fwqrc.wordpress.com&obj_id=151501655-490-653e422a30361Leave a commentGMP
What is E. coli?

E. coli are mostly harmless bacteria that live in the intestines of people and animals and contribute to intestinal health. However, eating or drinking food or water contaminated with certain types of E. coli can cause mild to severe gastrointestinal illness. Some types of pathogenic (illness-causing) E. coli, such as Shiga toxin-producing E. coli (STEC), can be life-threatening.
People infected with pathogenic E. coli can start to notice symptoms anywhere from a few days after consuming contaminated food or as much as nine days later. Generally, the symptoms include severe stomach cramps, diarrhea, fever, nausea, and/or vomiting.
The severity or presence of certain symptoms may depend on the type of pathogenic E. coli causing the infection. Some infections can cause severe bloody diarrhea and lead to life-threatening conditions, such as a type of kidney failure called hemolytic uremic syndrome (HUS), or the development of high blood pressure, chronic kidney disease, and neurologic problems. Other infections may have no symptoms or may resolve without medical treatment within five to seven days.
Due to the range in severity of illness, people should consult their health care provider if they suspect that they have developed symptoms that resemble an E. coli infection, including HUS, but even healthy older children and young adults can become seriously ill.
People of any age can become infected with pathogenic E. coli. Children under the age of 5 years, adults older than 65, and people with weakened immune systems are more likely to develop severe illness as a result of an E. coliinfection. However, even healthy older children and young adults can become seriously
Share this:
Loading…Leave a commentGMP
Outbreak Investigation of lllnesses caused by E. coli O157:H7 November 2019
FDA working to determine the source of contamination found in Ready Pac Bistro Chicken Caesar Salad, FSIS Announces Recall
On Nov. 21, 2019, the U.S. Department of Agriculture’s Food Safety and Inspection Service (FSIS) announced a recall by Missa Bay, LLC, a Swedesboro, N.J. establishment. The salad products subject to the recall can be found in a spreadsheeton the FSIS website and bear the USDA mark of inspection.
Do not eat the recalled products.
The salad product items were produced from Oct. 14, 2019 through Oct. 16, 2019, and the recommended “use by” date for all of the product was Nov. 1, 2019, or earlier. There is concern that some products may be in consumers’ refrigerators, or that portions may have been frozen, even though they are past their use by dates. These products should be thrown away or returned to the place of purchase.
Background
FDA, CDC, FSIS and state health authorities are investigating an outbreak of illnesses caused by E. coli O157:H7 in the U.S.

Products in the recall noted above were produced with the same lot of lettuce that was used to produce the packaged salad that the Maryland Department of Health found to contain E. coli 0157:H7.
That product was Ready Pac Bistro® Chicken Caesar Salad, lot #255406963, UPC 0 77745 27249 8, “Best By” date Oct. 31, 2019, and it is included in this recall.
FDA is tracing back the supply of the romaine lettuce in the Caesar salad. FDA has identified possible farms in Salinas, California. FDA is deploying investigators to determine the source and extent of the contamination. More information will be forthcoming as the investigation proceeds.
Although the ill people interviewed in Maryland reported eating Ready Pac Bistro® Chicken Caesar Salad, at this time, ill people in other states have not reported eating this particular salad. Therefore, exposure to this product alone does not fully explain other cases in the outbreak.
Case Counts
Total Illnesses: 17
Hospitalizations: 7
Deaths: 0
Last illness onset: November 8, 2019
States with Cases: AZ (1), CA (2), CO (1), ID (3), MD (2), MT (1), WA
Share this:
Loading…Leave a commentGMP
FDA APPROVES NEW TREATMENT FOR ADULTS WITH PARTIAL ONSET-SEIZURES
The U.S. Food and Drug Administration today approved XCOPRI (cenobamate tablets) to treat partial-onset seizures in adults“XCOPRI is a new option to treat adults with partial-onset seizures, which is an often difficult-to-control condition that can have a significant impact on patient quality of life,” said Billy Dunn, M.D., director of the Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research. “Patients can have different responses to the various seizure medicines that are available. This approval provides an additional needed treatment option for people with this condition.”
A seizure is a usually short episode of abnormal electrical activity in the brain. Seizures can cause uncontrolled movements, abnormal thinking or behavior, and abnormal sensations. Movements can be violent, and changes in consciousness can occur. Seizures occur when clusters of nerve cells (neurons) in the brain undergo uncontrolled activation. A partial-onset seizure begins in a limited area of the brain
The safety and efficacy of XCOPRI to treat partial-onset seizures was established in two randomized, double-blind, placebo-controlled studies that enrolled 655 adults. In these studies, patients had partial-onset seizures with or without secondary generalization for an average of approximately 24 years and median seizure frequency of 8.5 seizures per 28 days during an 8-week baseline period.
During the trials, doses of 100, 200, and 400 milligrams (mg) daily of XCOPRI reduced the percent of seizures per 28 days compared with the placebo group. The recommended maintenance dose of XCOPRI, following a titration (medication adjustment) period, is 200 mg daily; however, some patients may need an additional titration to 400 mg daily, the maximum recommended dose, based on their clinical response and tolerability
Share this:
Loading…Leave a commentGMP
FDA, CDC, and state health authorities are investigating an outbreak of illnesses caused by E. coli O157: H7 in the U.S.
Regulatory focus news letter

According to the CDC, as of November 18, 2019, 17 people infected with the outbreak strain of E. coli O157:H7 have been reported from eight states. The case patients report that illnesses started on dates ranging from September 24, 2019 to November 8, 2019.
Two cases reported from Maryland have been linked to this outbreak by Whole Genome Sequencing (WGS), through analysis of clinical samples taken from those patients. The Maryland Department of Health identified E. coli O157 in an unopened package of Ready Pac Bistro® Chicken Caesar Salad collected from a ill person’s home in Maryland which was purchased from a Sam’s Club in that state. Preliminary information indicates that romaine lettuce used in the product that tested positive was harvested in mid-October and is no longer within current expiration dates. To date, the food sample has not yet been definitively linked to the Maryland cases or other E. coli O157 illnesses in the multi-state outbreak. WGS analysis is currently underway for this sample to determine if it is closely related genetically to the E. coli found in people in this outbreak.
As analysis is underway, FDA is tracing back the supply of the romaine lettuce in the Caesar salad. FDA has identified possible farms in Salinas, California. FDA is deploying investigators to the farms in question to determine the source and extent of the contamination. More information will be forthcoming as the investigation proceeds.
Although the ill people interviewed in Maryland reported eating Ready Pac Bistro® Chicken Caesar Salad, at this time, ill people in other states have not reported eating this particular salad. Therefore, exposure to this product alone does not fully explain other cases in the outbreak.
Share this:
Loading…Leave a commentFWQRC, GMP, LIFE SCIENCES, PHARMA
Need for a proactive approach-To protect our loved one and also to continue the business without financial drop down.
We have heard the presence of NDMA impurities in Sartans and Ranitidine drugs. Initially it was only sartans, later Ranitidine and what’s next now?
Now let’s extend the concept to OTC drugs that may contain Nitrosamines impurity. Lets us review the sources of Nitrosoamines.
- Solvents:
The source for nitroso amines may also be from solvents.
DMF: NDMA can also be formed from Dimethyl formamide as shown below.

DMA is present as an impurity in DMF, a precursor in the industrial DMF process. It may also formed as a degradant during storage of the solvent.
DEA:

Similar to DMA formation, DEA could be formed by degradation of triethylamine (TEA) or exist as impurity in TEA raw material.
Some common organic solvents (e.g. NMP which could give rise to 4- (methyl)(nitroso)amino)butanoic acid = NMBA) and amine bases (e.g. diisopropylamine = DIPEA which could give rise to N-Nitrosodiisopropylamine (DIPNA) and N-Nitrosoethylisopropylamine (EIPNA)) would present such risks.
- Reagents Like piperazine, a secondary amine, reacts slowly with NOx in the presence of O2 to form a nitrosamine derivative under conditions similar to those found in industrial amine-based post-combustion CO2 capture processes.
The genotoxic and mutagenic assessment is at most performed for Raw materials, reagents, catalysts etc., But do we really assess the Recycled solvents, reagents and catalysts and this may be chance for nitrosamine formation due to the presence of amines in the waste streams sent for recovery and the subsequent quenching of these materials with nitrous acid to destroy residual azide, without adequate control of nitrosamine formation or adequate purification.
Examples of recycled materials observed to be contaminated with nitrosamines include orthoxylene and tributyltin chloride (used as a source of tributyltin azide). Nitrosamines may be entrained if they have similar boiling points or solubility properties to recovered materials depending on how recovery and subsequent purification takes place (e.g. aqueous washes or distillation).
Simple checks during the manufacturing process can identify the impurities and avoids the market recall.
- Evaluate the solvents, reagents for the structural alert that are similar like nitrosoamines.
- Recovered solvents, recovered materials that are outsourced by a third party vendor.
- Evaluation of drug product formulation and process.
In-depth analysis during drug development should actually identify all the issues.
Budding and Small-scale industries who cannot identify the sources of the impurity or who cannot have sufficient capacity to analyse the drugs can take the help of qualified labs/ consultancies as a onetime activity.
Best advice is our cost saving should not cost the life of our fellow citizens.
A proactive approach for the high market value molecules to avoid any surprise in the future.
OTC Drugs
- Drugs that contains Piperazine ring are Ranolazine, Trimetazidine, Amoxapine, Amoxapine, Befuraline, Buspirone, Flesinoxan, Ipsapirone, Nefazodone, Piberaline, Tandospirone, Trazodone, Vilazodone, Zalospirone, Meclozine, Cinnarizine, Hydroxyzine, Cetrizine, Levo citrizine, Niaprazine, Fluphenazine, PERPHENAZINE, Prochlorperazine, THIOTHIXENE, Quipazine, Imatinib, Benzylpiperazine, Buclizine, Ziprasidone,Etc…
Out of the above drugs listed, The OTC drugs are Cetirizine, Hydroxyzine etc are into the consideration of Nitrosoamine impurities and other prescription should also be taken into consideration.
- One of the article which was published in the year 2001, has concluded that nitrosoamine was detected in the patients urine, who is consuming the long term treatment with Omeprazole, a Proton pump inhibitor.
Omeprazole, with the maximum daily dose of 360 mg/day is prescribed for some patients with Zollinger-Ellison Syndrome for treatment period longer than 5 years.
Since, these OTC drugs are easily available in the market and consumed without doctors prescription, there is a quick need for evaluation and control of these impurities in the Drug substance as well as drug product.
The above statement is just an example and further evaluation is REQUIRED.
Since the method for the detection of the nitrosamines is available, why delay, start assessing and perform a risk assessment for your drugs.
Assess, Analyse, Announce your results and be responsible for public health, since these are OTC drugs.
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=364&origin=fwqrc.wordpress.com&obj_id=151501655-364-653e422ea8563Tagged DMF, Genotoxicity, NDMA, Nitrosoamines, Risk AssessmentLeave a commentFWQRC, GMP, LIFE SCIENCES, PHARMA
NDMA-Genotoxic Impurity

Hello Readers!!!
Today’s discussion is about the hottest topic in the pharmaceutical industry, where many industries producing the Ranitidine and Valsartan drugs recalled the batches from the MARKET due to the presence of this impurity in the drug above the acceptance criteria.
First of all, What is NMDA and NDEA impurity?
N-Nitrosodimethylamine (NDMA), also known as dimethylnitrosamine (DMN) and N-nitrosodiethylamine, the member N-nitrosoamine class is a semi-volatile organic chemical, produced as a byproduct in chemical synthesis or as a whole.
A bit more about NDMA & NDEA impurities.
NDMA

NDEA

Numerous studies were conducted in vitro in bacterial and mammalian cells, there has been overwhelming evidence that NDMA is not only a mutagenic but also clastogenic. The NDMA impurity increased the frequencies of gene mutations, chromosomal damage, sister chromatid exchange, and unscheduled DNA synthesis in a wide variety of cell types including human and rodent cells.
The evidence of genetic effects has also been observed in in vivo studies. Clastogenic effects (e.g., micronuclei, sister chromatid exchange, chromosomal aberrations) in, bone marrow cells, spleen cells and peripheral blood lymphocytes, as well as in oesophageal, kidney cells have been observed in rodents (rats, mice, or hamsters) administered NDMA either orally or by intraperitoneal injection.
Evidence of genotoxicity (e.g., chromosomal aberrations, micronuclei, gene mutation,DNA strand breaks) has also been observed in the offspring of hamsters and mice.
How does it reacts with the human body?
NDMA is metabolized by CYP2E1 enzyme in the liver, which hydroxylates one methyl group. The resulting hydroxymethyl nitrosamine is unstable and decomposes to formaldehyde, which is also used to quantify the metabolic rate and methane-diazonium-ion, which methylates DNA and protein or reacts with water to methanol. The electrophilic intermediates produced by metabolic activation of nitrosamines, react rapidly with cellular nucleophiles and target for carcinogens during tumor initiation.

The discussion about the NDMA impurities has been conversed previously.
- In ICH M4 guideline “ASSESSMENT AND CONTROL OF DNA REACTIVE (MUTAGENIC) IMPURITIES IN PHARMACEUTICALS TO LIMIT POTENTIAL CARCINOGENIC RISK”- Dated March 31, 2017.
- World Health Organization Guidelines for Drinking-Water Quality, 3rd edition including 1st and 2nd addenda, 2008.
The topic is not only limited to Ranitidine and Valsartan drugs but it is applicable to all the drugs. All the drug substances should be assessed for the NDMA impurity and necessary controls should be taken.
Lets not wait for the regulatory authorities to take action upon the manufacturers. It is the Pharmaceutical manufacturers responsibility to deliver the right drugs to ensure the patient safety…
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=297&origin=fwqrc.wordpress.com&obj_id=151501655-297-653e42329c34cTagged API, Genotoxicity, IMPURITIES, NDMA, PHARMACEUTICAL MANUFACTUERESLeave a commentGMP
Business Incubators
As we know, Indian Incubators are growing swiftly and they offer a great support to the entry level entrepreneurs across India. But how talented the start-up’s are they need some external help. Because it takes some time to grow from seed level to a plant. These Indian Incubators form a perfect bridge from idea to execution. They nurture young firms and help them to survive in the early stage. Incubators are offered by non -profit organizations like government, business alliance etc.
Here are some special features of Incubators
- Tackle burdensome regulations
- They enable entrepreneurs to learn from each other
- They also promote cultural change and economic growth
- They offer one-stop facility for counsel, skills, shared facilities
- They provide networking to mobilize external services and mentoring
How to choose incubator
There are four main things to choose the right incubator
- First select the program type in which you want to enroll. i.e. accelerator (or) incubator
Incubators will nurture a business in its startup phase and allow it to develop at its own pace making them ideal for entrepreneurs who want to grow their company steadily over time.
- Second is location
It is better to select a program type that is located close to your city, in order to have more interaction with your mentors, partners etc. It is beneficial later. And also certain cities may be better for particular industries. Like New York for media, Los Angeles for entertainment etc,.
- The next is Industry focus, is another element yet to consider. You need to check the industry the incubator or accelerator specializes in. you need to choose an incubator that connect you to leaders in your industry, and also provide industry specific facilities.
- And finally, services that are provided by incubator. Make sure the services offered by incubators are completely desired. You need to find the one that offer necessary services for your startup.

Image source: https://media1.picsearch.com/is?gpc0LlhBtU-2axkHhvkdANipP-KeWULjKOxQlbra_ao&height=272
First of all, make a list of your desired services first and then wanting your options accordingly.
TOP INSTITUTE STARTUP INCUBATORS IN INDIA
- TBI, BITS Pilani.
- TBI, VIT Vellore.
- RTBI, IIT Madras.
- SINE, IIT Bombay.
- CIIE, IIM Ahmadabad.
- SIDBI SIIC, IIT Kanpur.
- TREC STEP, NIT Trichy.
- NSRCEL, IIM Bangalore.
TOP STARTUP INCUBATORS IN INDIA
- iCreate
- Indavest
- Seed fund
- Angel Prime
- Khosla Labs
- Indian Angel Network
- Amity Innovation Incubator
- Science and Technology Entrepreneurship Park, -IITK
- Centre for Innovation Incubation and Entrepreneurship -IIMA
- Nadathur S Raghavan Centre for Entrepreneurial Learning (NSRCEL)
Author : Muthamizh selvi
Share this:
Loading…Leave a comment
QUALITY RISK MANAGEMENT
Hi Everyone
Good evening……..
Today we are going to discuss about Quality Risk Management process
Since a couple of years Quality Risk Management (QRM) has become a mandatory regulatory requirement towards healthcare organizations.
QRM is an overall and continuing process of minimizing risks to product quality throughout its life-cycle in order to optimize its benefit and balance the risk.
It is a systematic process for the evaluation, control, communication and review of risks to the quality of the medicinal product.
It supports science based and practical decisions when integrated into quality systems, examples of quality systems include Validation, Quality Defects – Investigation, Auditing, Inspection, Documentation, Training etc.
Quality Risk Management principles are effectively utilized in many areas including business, insurance, work related safety, public health, pharmacovigilance, and by agencies regulating these industries.
Even though there are some examples of the use of quality risk management in the pharmaceutical industry, today they are limited and do not represent the full contributions that risk management has to offer.
In relation to pharmaceuticals, though there are a variety of stakeholders, including medical practitioners and patients as well as government and industry, the safety of the patient by managing the risk to quality should be considered prime importance.
The manufacturing and use of a drug product, including its components, necessarily involve some degree of risk.
An effective QRM approach can further ensure the high quality of the drug product to the patient by identify and control potential quality issues during development and manufacturing.
Use of QRM can improve the decision making if a quality problem arises. Effective QRM implementation can facilitate better and well- versed decisions which can provide regulators with greater assurance of a company’s ability to deal with possible risks.
Principles of Quality Risk Management
Four primary principles of QRM are:
- The assessment of the risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient.
- QRM should be dynamic, iterative and responsive to change.
- The level of effort, formality and documentation of the QRM process should be commensurate with the level of risk
- The capability for continual development and enhancement should be embedded in the QRM process.
General Quality Risk Management Process

Quality Risk Management is a systematic process for evaluation, control, communication and review of risks to the quality of the drug product across the product life cycle.
Risk can be defined as the combination of the probability of occurrence of harm and the severity of that harm
Initiating a Quality Risk Management Process
Quality Risk Management should include systematic processes designed to organise, facilitate and improve science-based decision making with respect to risk. Steps used to initiate and plan a quality risk management process might include the following:
- Define the problem and/or risk question, including relevant assumptions identify the potential for risk.
- Assemble background information and/or data on the potential hazard, harm or human health impact applicable to the risk assessment.
- Specify a timeline, and appropriate level of decision making for the risk management process.
Risk Assessment
Risk assessment consists of the identification of hazards and the analysis and evaluation of risks associated with exposure to those hazards.
It includes risk identification, risk analysis and risk evaluation. Three fundamental questions are often helpful.
- What might go wrong?
- What is the possibility that it will go wrong?
- What are the consequences?
Risk identification
Risk identification is a organized use of information to identify hazards referring to the risk.
Information can include historical data, theoretical analysis, and the concerns of stakeholders.
Risk identification addresses the “What might go wrong?” question, including identifying the possible consequences.
This provides the basis for further steps in the quality risk management process.
Risk analysis
Risk analysis is the estimation of the risk associated with the identified hazards.
It is the qualitative or quantitative process of linking the likelihood of occurrence and severity of harms.
In some risk management tools, the ability to detect the harm (detectability) also factors in the estimation of risk.
Risk evaluation
Risk evaluation compares the identified and analysed risk against given risk criteria.
Risk evaluations consider the strength of evidence for all three of the fundamental questions.
Different Steps Involved In the Risk Assessment Are
- Collect & organise the information
- Formulate the Risk Question
- Choose Tool different tools include
- Identify Risks Factors and Related Hazards
- Define the Risk Components &Scales
- Evaluate the risk for each hazard
- Determine acceptability of risks
- Determine Action Threshold
- Apply the tool
Risk control
Risk control includes decision making to reduce and/or accept risks.
The intention of risk control is to reduce the risk to an acceptable level.
The amount of effort used for risk control should be proportional to the significance of the risk
Risk reduction focuses on processes for mitigation or avoidance of quality risk when it exceeds a specified level.
Risk reduction might include actions taken to mitigate the severity and probability of harm.
The implementation of risk reduction measures can introduce new risks into the system or increase the significance of other existing risks.
Hence, it might be appropriate to revisit the risk assessment to identify and evaluate any possible change in risk after implementing a risk reduction process.
Risk acceptance is a decision to accept risk.
For some types of harms, even the best quality risk management practices might not entirely eliminate risk.
In these circumstances, it might be agreed that an appropriate quality risk management strategy has been applied and that quality risk is reduced to a specified (acceptable) level.
This (specified) acceptable level will depend on many parameters and should be decided on a case-by-case basis.
Risk Review is the output/results of the risk management process should be reviewed to take into account new knowledge and experience.
Once a quality risk management process has been initiated, that process should continue to be utilized for events that might impact the original quality risk management decision.
Risk review might include reconsideration of risk acceptance decisions
Risk Communication is the sharing of information about risk and risk management between the decision makers and others.
The output/result of the quality risk management process should be appropriately communicated and documented.
The included information might relate to the existence, nature, form, probability, severity, acceptability, control, treatment, detectability or other aspects of risks to quality.
Conclusion:
Quality Risk Management is a systematic process for evaluation, control, communication and review of risks to the quality of the drug product across the product lifecycle.
Effective Quality Risk Management can facilitate better and more informed decisions, can provide regulators with greater assurance of a company’s ability to deal with potential risks, and might affect the extent and level of direct regulatory oversight.
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=181&origin=fwqrc.wordpress.com&obj_id=151501655-181-653e4232a6ba2Leave a comment

WHAT IS COMMON TECHNICAL DOCUMENT?
Hi Everyone
Good evening………………………
Today We are going to discuss on eCTD
CTD is an ICH standard that FDA adopted in a consensus process, as a member of ICH, together with other member regions, Europe and Japan Currently global format for regulatory submissions Consistent data organization Method to electronically transfer product information and data Collection of electronic files organized according to guidelines defining file format, folder/files naming convention, document specifications etc. Applies to all NDAs, ANDAs, BLAs, INDs and master files.

The eCTD challenge In the US, eCTD-only NDAs, BLAs and INDs are accepted – no paper necessary eCTD plus paper still needed for Medical authorities in EU Paper is still the official archival copy of the EU MA EU wants eCTD as preferred format for all Marketing Authorisation Applications(MAAs) and variations Only eCTD for MA for all EU members states by 1 Jan 2010 Health Canada wants eCTD format on CD/DVD plus paper
NEED FOR ELECTRONIC SUBMISSIONS
Designed with consideration that facilitate Creation Review Assists project management and information management Life cycle management (the history of a product application) Archiving Drug development planning
Current Status of US eCTD Submissions FDA Office of Chief Information Officer Quarterly briefing, 12 Dec 2008 During the period 2005 to 2008, eCTD submission volume grew at a compounded annual growth rate of approximately 300%.
Basics of eCTD The ICH M4 Expert Working Group (EWG) has defined the Common Technical Document (CTD). The ICH M2 EWG has defined, in the current document, the specification for the Electronic Common Technical Document (eCTD). There are 5 modules in the submission report. A hierarchical cabinet/folder structure containing the electronic documents (PDF). An XML backbone that provides a structure to display the PDF documents in the eCTD format (eCTD viewer)
References
- CDER Contact for information on eCTD submissions esub@fda.hhs.gov
- CDER Contact for information on SDTM submission cder-edata@fda.hhs.gov Electronic
- Regulatory Submissions and Review website http://www.fda.gov/cder/regulatory/ersr/default.htm
- International Conference on Harmonization http://www.ich.org
- All FDA Guidances on Electronic Submissions http://www.fda.gov/cder/guidance/index.htm#electronic_submissions
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=177&origin=fwqrc.wordpress.com&obj_id=151501655-177-653e4232aaf5c4 CommentsGMP
Data Integrity Issues & Concerns
Good evening everyone………..
Today we are going to discuss on Data integrity issues
What is Data Integrity?
- Complete, consistent, and accurate data to assure patient safety and product quality.
- Complete, consistent, and accurate data should be attributable, legible, contemporaneously recorded,original or a ‘true copy’, and accurate (ALCOA).
- Good Documentation Practices for Static and Dynamic Records.
- Data integrity should be maintained throughout the data life cycle, including, but not limited to data creation, processing, archiving and disposition after record’s retention period

What is ALCOA?
- Attributable – Traceable to a unique individual
- Legible – Data must be recorded permanently and be readable
- Contemporaneously – Activities must be recorded at the time they occur
- Original or a true copy – first capture of data (not transcribed
data), must review the original record, must retain the original or certified copy of the original record. - Accurate – records must be accurate, which is achieved thru the Quality Management System
What are the Regulations?
- Data in accordance with cGMP requirements for drugs (i.e., as required by 21 CFR parts 210, 211,and 212).
- Part 210 – Current Good Manufacturing Practice in Manufacturing,Processing,
- Packing, or Holding of Drugs; General.
- Part 211 – Current Good Manufacturing Practice for Finished Pharmaceuticals.
- Part 212 – Current Good Manufacturing Practice for Positron Emission Tomography Drugs.
- Q7A – Active Pharmaceutical Ingredients.
Regulations – Data Integrity : Requirements with respect to data integrity in parts 211 and 212 continued:
- 211.188, 211.194, and 212.60(g) (requires “complete information,” “complete data derived from all tests,” “complete record of all data,” “original records have been reviewed for accuracy, completeness, and compliance with established standards,” and “complete records of all tests performed”).
- 211.192 (requires production and control records be “reviewed”) 211.101(c) and (d), 211.103, 211.182, 211.186(a), 211.188(b)(11), and 211.194(a)(8) require records be “reviewed” by a second person.
Why is Data Integrity Important?
- FDA cGMP inspection(s) have uncovered violations with data integrity issues.
- Data integrity is an important component of industry’s responsibility to ensure the safety, efficacy, and quality of drugs, and of FDA’s ability to protect the public health.
- Data integrity-related cGMP violations may lead to regulatory actions, including warning letters, import alerts, and consent decrees.
- The underlying premise in 210.1 and 212.2 is that cGMPs sets forth minimum requirements to assure drugs meet standards of the Federal Food, Drug, and Cosmetic Act (FD&C Act) regarding safety, identity, strength, quality, and
purity. - FDA’s authority for cGMP comes from FD&C Act section 501(a)(2)(B).
- 501(a)(2)(B) states: a drug shall be deemed adulterated if “the methods used in, or the facilities or controls used for, its manufacture, processing, packing, or holding do not conform to or are not operated or administered in conformity with current good manufacturing practice to assure that such drug meets the requirement of the act as to safety and has the identity and strength, and meets the quality and purity characteristics, which it purports or is represented to possess.”
- Reliability on the information used to ensure the quality of the drugs that
consumers will take - Data integrity problems break trust
- FDA rely on firm’s to do the right thing when FDA is not present.
Examples of significant issues
- No raw data to support records
- Creating inaccurate and incomplete records
- Test results for one batch used to release other batches
- Backdating
- Fabricating data
- Discarding data
- Repeated tests, trial runs, sample runs (testing into compliance)
- Changing integration parameters of chromatographic data to obtain passing results
- Deletion/manipulation of electronic records
- Turning off audit trail
- Sharing password
- Inadequate controls for access privileges
- Inadequate/incomplete computer validation
- Inadequate investigations
- Inaccurate reporting of microbial, sterility, or endotoxin data results
- Loss of data during changes to the system
- Activities not recorded contemporaneously
- Employees that sign that they completed manufacturing steps when the employees were not on premises at the time the steps were completed.
Things to consider…
- Is data integrity a problem at your facility? What measures are in place to prevent data integrity problems?
- Are internal audit procedures adequate? What measures are in place and will they detect data integrity issues?
- Does senior management cultivate adequate and accurate reporting of events when things go wrong during manufacturing…during testing?
- Train personnel to detect and prevent data integrity as part of routine cGMP training.
Where does the Agency find Data Integrity Issues?
- Domestic and international facilities
- Small and large pharmaceuticals companies
- Manufacturing operations
- Quality units, including quality control laboratories (chemistry and microbiology)
- Clinical Trials
Things to consider…
- Existing systems should be able to ensure data integrity, traceability and reliability
- Firms who outsource operations should have robust systems in place to verify and compare data generated by the contractor
Conclusion
- Once data integrity issues are found during an inspection, a change to a written
procedure or firing an employee is not enough - Quality Risk Management approaches to prevent, detect and control potential risk
are essential
FDA recommends that data integrity problems identified during inspections
be addressed?
- The firm should demonstrate effective remediation. For example, By:
- Hire a third party auditor
- Determine the extent/scope of the problem
- Implement a global corrective action plan
- Removing individuals responsible for problems from cGMP positions
References
- Part 210 – Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding of Drugs; General.
- Part 211 – Current Good Manufacturing Practice for Finished Pharmaceuticals.
- Part 212 – Current Good Manufacturing Practice for Positron Emission Tomography Drugs.
- Q7A – Active Pharmaceutical Ingredients.
Thank you………..
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=103&origin=fwqrc.wordpress.com&obj_id=151501655-103-653e4232ad39a2 CommentsGMP
CEP/COS-TOP 10 DEFICIENCIES

Before starting the discussion about the deficiencies that we receive from the agencies…
Let me ask a question to all of the readers!
Have you received a CEP certificate without a single deficiency letter or request for additional information?

The answer might be big “NO”.
So the today article mainly focuses on the top 10 deficiencies that we frequently receive from the regulatory authorities or agencies.
This article is in conjugation with PA/PH/CEP (16) 58 document, discussed to avoid the receipt of the deficiency letters. Because the management of CEP application policy allows only two deficiency letters to be issued to the manufacturing companies. If the dossier is still considered deficient the CEP application may be rejected.
So, by following the below checks, which are listed in the top 10 deficiencies in the CEP PA/PH/CEP (16) 58 document, while preparing the dossier may avoid the queries in the deficiency letter.
Number 1: Genotoxic/ Mutagenic impurities
The CEP applications which are being filled by the manufacturing companies are lacking the discussion on Genotoxic and mutagenic impurities. Identifying the compounds that possess a structural alert and controlling the compounds below the TTC level could be the solution.
- Perform the genotoxic assessment of the Active pharmaceutical ingredient right from the Route of synthesis of the starting material.
- Calculate the TTC value and identify a suitable analytical method to show the absence or control measure in the final drug substance.
Number 2: Choose a suitable starting material
The 50% of the pharmaceutical companies receive this most common deficiency for choosing a suitable starting material for the API.
- The starting material should be choosed based on the ICH Q11 guideline.
- The starting material should not possess a complex structure and it should not have a major portion of the API.
Number 3: Inadequate discussion of the manufacturing process.
The regulatory agencies look for the detailed information about the manufacturing process and the API manufacturers should completely provide the documents related to the manufacturing process.
- Recovery processes and Recovery solvents batch processing records are expected to be present in the section 3.2.S.2.2.
- Batch size mentioned in the section 3.2.S.2.2 should match with the Batch analysis section 3.2.S.4.4.
Number 4: Carryover of starting material impurities to the final drug substances
Choosing the suitable starting material is a task, and setting a proper specification to the starting material is another task.
- Most of the starting materials may be isomers, so controlling the relative isomers in the starting material is one major check. In the current scenario, the regulatory agency asks the manufacturer to show the absence of isomers in the final drug substance.
- The specification of the starting material should have an identification test, limit for specified, unspecified and total impurities.
Number 5: Improper Justification of the starting material specification
Even though the starting material specification is set with the limits with specified, unspecified and total impurities, without proper justification for set limits, the agency could not accept the specification and hence leads to the deficiency letter.
- The justification of the specification should include the evaluation of the risks and the ability of the subsequent steps to purge impurities.
- Technical write up for the process to purge the impurities should be included in the dossier.
Number 6: Inadequate specifications for the solvents (recycled and recovered) used in the manufacturing process.
The usage of recovered solvents is not restricted in the manufacturing process but the specification should be set and testing should be performed to prove that the use of recovered solvents does not affect the quality of the drug substance.
- The set specification should include a purity test and the reasonable mass balance is expected to be in place.
Number 7: Inadequate specifications for the reagents and elemental impurities
The manufacturing process may or may not use metals, but the manufacturing process including equipment, utilities should be screened for the presence of elemental impurities. The ICH Q3D guideline defines the elemental impurities and divided the elements into three classes namely 1, 2A, 2B and Class 3. The regulatory agencies expects a risk assessment summary for the 24 elements.
- Provide a risk assessment for all the 24 elements in the dossier section 3.2.S.3.2.
- Elements such as Boron, aluminium etc if used in the synthesis of the drug substance, a discussion about their absence in the final drug substances is expected.
Number 8: Inadequate specifications for the isolated intermediates
It is presumed that the specification of the intermediates should be set in such a way that it captures all the related impurities in the intermediate specification.
Number 9: Justification and carryover of isolated impurities
The impurities captured in the intermediate specification should have definite limit, justification is supposed to the set limit and if any impurity limit in the intermediate is set more than 1.0%, then it absence in the final drug substance should be shown. The risks of having uncontrolled impurities in the final substance potentially above acceptable limits should be addressed
Number 10: In adequate information about the starting materials.
The following information about the starting materials is necessary to be present in the dossier.
- Route of synthesis
- Address of the manufacturer
- Brief description with flow chart.
If more than one source of starting material is used in the process, batch equivalency report is to be provided in the dossier.
Avoiding the above checks could bring us the quick approval.
ALL THE BEST
References:
Share this:
Loading…4 CommentsGMP
How to Plan and Implement an Effective Corrective Action System
Hi Everyone
Good evening…………
Today we are going to discuss on the purpose of a quality management system.
QMS is to help businesses improve abilities to consistently meet customer or regulatory requirements, according to the American Society for Quality.
A major component of a successful system is a corrective action program that adequately addresses non-conformance. The problem is that many systems become a burden rather than a tool for improvement, generally, because they are not well-planned prior to implementation.
CAPA Process
- Root cause investigation
- Conclusion & Quality design
- Action plan
- Implementation and follow up
Important Considerations
- Tie CAPA implementation to:
- Document control for products and processes
- Change control
- Ensure that controlled documents are reviewed and approved if changes are made.
NOTE: Closing CAPAs when actions are implemented, and tracking the effectiveness checks for CAPAs as a separate quality system metric. If closure takes more than 90 days, the CAPA should probably be converted to a Quality Plan. This is NOT intended to be a “work around” to give companies a way to extend CAPAs that are not making progress in a timely manner.
Share this:
Loading…2 CommentsPHARMA
Genotoxic Impurities
Hi Everyone…
Good evening
Today we are going to discuss on the Genotoxic Impurities and its assessments.
The toxicological assessment of genotoxic impurities and the determination of acceptable limits for such impurities in active substances is a difficult issue.
The data set usually available for genotoxic impurities is quite variable and is the main factor that dictates the process used for the assessment of acceptable limits.
In the absence of data usually needed for the application of one of the established risk assessment methods, i.e. data from carcinogenicity long-term studies or data providing evidence for a threshold mechanism of genotoxicity, implementation of a generally applicable approach as defined by the Threshold of Toxicological Concern (TTC) is proposed.
A TTC value of 1.5 µg/day intake of a genotoxic impurity is considered to be associated with an acceptable risk (excess cancer risk of <1 in 100,000 over a lifetime) for most pharmaceuticals.
From this threshold value, a permitted level in the active substance can be calculated based on the expected daily dose. Higher limits may be justified under certain conditions such as short-term exposure periods.
For determination of acceptable levels of exposure to genotoxic carcinogens considerations of possible mechanisms of action and of the dose-response relationship are important components. Based on the above considerations genotoxic impurities may be distinguished into the following two classes:
- Genotoxic compounds with sufficient (experimental) evidence for a threshold-related mechanism
This approach calculates a “Permitted Daily Exposure” (PDE), which is derived from the NOEL, or the lowest- observed effect level (LOEL) in the most relevant (animal) study using “uncertainty factors” (UF).
- Genotoxic compounds without sufficient (experimental) evidence for a threshold-related mechanism
The assessment of acceptability of genotoxic impurities for which no threshold mechanisms are identified should include both pharmaceutical and toxicological evaluations. In general, pharmaceutical measurements should be guided by a policy of controlling levels to “as low as reasonably practicable” (ALARP principle), where avoiding is not possible. Levels considered being consistent with the ALARP principle following pharmaceutical assessment should be assessed for acceptability from a toxicological point of view.
A TTC value higher than 1.5 µg/day may be acceptable under certain conditions, e.g. short-term exposure, for treatment of a life-threatening condition, when life expectancy is less than 5 years, or where the impurity is a known substance and human exposure will be much greater from other sources (e.g. food). Genotoxic impurities that are also significant metabolites may be assessed based on the acceptability of the metabolites.
The concentration limits in ppm of genotoxic impurity in drug substance derived from the TTC can be calculated based on the expected daily dose to the patient using equation (1).
(1) Concentration limit (ppm) = TTC [µg/day]/dose (g/day]
The TTC concept should not be applied to carcinogens where adequate toxicity data (long-term studies) are available and allow for a compound-specific risk assessment.
REFERENCES
- Cheeseman M.A., Machuga E.J., Bailey A.B., A tiered approach to threshold of regulation, Food Chem Toxicology 37, 387-412, 1999
- Dobo K.L., Greene N., Cyr M.O., Caron S., Ku W.W., The application of structure-based assessment to support safety and chemistry diligence to manage genotoxic impurities in active pharmaceutical ingredients during drug development, Reg Tox Pharm 44, 282-293, 2006.
- Gold L.S., Sawyer C.B., Magaw R., Backman G.M., de Veciana M., Levinson R., Hooper N.K., Havender W.R., Bernstein L., Peto R., Pike M.C., Ames B.N., A carcinogenic potency database of the standardized results of animal bioassays, Environ Health Perspect 58, 9-319, 1984.
- Kroes R., Renwick A.G., Cheeseman M., Kleiner J., Mangelsdorf I., Piersma A., Schilter B., Schlatter J., van Schothorst F., Vos J.G., Würtzen G., Structure-based threshold of toxicological concern (TTC): guidance for application to substances present at low levels in the diet, Food Chem Toxicol 42, 65-83,2004.
- Kroes R., Kozianowski G., Threshold of toxicological concern (TTC) in food safety assessment, Toxicol Letters 127, 43-46, 2002.
- Müller L., Mauthe R.J., Riley C.M., Andino M.M., De Antonis D., Beels C., DeGeorge J., De Knaep A.G.M., Ellison D., Fagerland J.A., Frank R., Fritschel B., Galloway S., Harpur E., Humfrey C.D.N., Jacks A.S.J., Jagota N., Mackinnon J., Mohan G., Ness D.K., O’Donovan M.R., Smith M.D., Vudathala G., Yotti L., A rationale for determining, testing, and controlling specific impurities in pharmaceuticals that possess potential for genotoxicity, Reg Tox Pharm 44, 198-211, 2006
- Munro I.C., Safety assessment procedures for indirect food additives: an overview. Report of a workshop. Reg Tox Pharm 12, 2-12, 1990.
- Munro I.C., Kennepohl E., Kroes R., A procedure for the safety evaluation of flavouring substances, Food Chem Toxicology 37, 207-232, 1999.
- Rulis A.M., Establishing a threshold of regulation. In Risk Assessment in Setting National Priorities(J.J. Bonin and D.E. Stevenson, Eds.) Plenum, New York, 271-278, 1989.
- U.S. Food and Drug Administration (FDA), Food additives: Threshold of regulation for substances used in food-contact articles (final rule), Fed. Regist. 60, 36582-36596, 1995.
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=20&origin=fwqrc.wordpress.com&obj_id=151501655-20-653e423aaafd2Tagged Genetics, Genotoxicity, Mutations Genes, Threshold level, TTCLeave a commentGMP, PHARMA
Root Cause Analysis for Drugmakers
Hi Everyone
Whenever regulatory authorities anywhere in the world perform an audit of a drug manufacturer, one of their most frequent findings remains the inadequate performance of the investigation of deviations.
Authorities expect stakeholders will carefully investigate deviations to identify non-compliance, intervene and then evaluate the effectiveness of that intervention. Without adequate investigation and root cause analysis (RCA), those stakeholders cannot effectively identify and design successful interventions. In fact, organizations waste millions of dollars every year on ineffective interventions.
A tool such as RCA uses a defined critical analysis approach in evaluating the reason for a deviation or non-conformance.
RCA techniques include brain storming, the “5 whys” and the “fishbone diagram.” Any and all may be used to explore and further examine the causes behind an event. Firms may then use the resulting analysis to identify areas for change, as well as any recommendations and solutions that aim to minimize the likelihood of an event repeating in the future.
Some organizations go so far as to create a corrective and preventive action (CAPA) for every event, although it is not always necessary.
A firm’s primary goals in investigating an incident should include both discovering its cause and ensuring it does not reoccur.
Regulatory authorities place a high value on RCA and CAPA. Indeed, a large number of FDA observations cite inadequate RCA, ineffective investigation and inappropriate CAPA.
When a deviation occurs, whether in the pharmaceutical or any other industry, the responsible firm must undertake an investigation to determine what went wrong and what damage, if any, the product might have suffered. The investigation process should include some specific steps. These include:
- Notification of the appropriate stakeholders;
- Containment action;
- Classification of the event;
- Decision to investigate;
- Determination of the root cause or RCA; and
- Review and approval process.
The effectiveness of the CAPA taken by an organisation marks another key step in the overall RCA. By monitoring the CAPA, an investigative team can determine whether it truly identified an incident’s root cause and then applied the “appropriate fix.”
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=30&origin=fwqrc.wordpress.com&obj_id=151501655-30-653e423aac30a1 CommentGMP
Temperature Mapping of Storage Areas
Hi everyone
Good evening
Today we are going to discuss how to carry out a systematic mapping procedure in any cold room, freezer rooms or other temperature-controlled store
1.0. Requirements:
All new temperature-controlled storage areas must be temperature-mapped as part of a fully documented verification process, before the installation is commissioned and handed over by the installer.
2.0. The temperature mapping procedures should:
Demonstrate the air temperature profile throughout the storage area, when empty and in a normal loaded condition;
- Define zones which should not be used for storage of TTSPPs “Time and temperature sensitive pharmaceutical product” (for example areas in close proximity to cooling coils, cold air streams or heat sources); andIf required, demonstrate the time taken for temperatures to exceed the designated limits in the event of power failure.
- Depending upon the routine monitoring strategy, subsequent mapping exercises may also be required on a periodic basis – for example, every three years – in order to demonstrate continuing compliance.
- All mapping exercises should be fully documented in order to demonstrate compliance to management, clients and the regulatory authorities.
3.0. A temperature mapping exercise involves a four stage process, as follows:
- Prepare a mapping protocol.
- Carry out the mapping exercise
- Prepare a mapping report.
- Implement the recommendations by carrying out the remedial and other actions identified in the mapping report. A follow-up mapping exercise may then be needed to verify the effectiveness of the remedial actions.
3.1. The mapping protocol
A detailed and comprehensive protocol should be prepared, reviewed and approved before the mapping exercise begins. A well-designed protocol will help ensure that the mapping study is correctly carried out. With suitable adjustments or options to cover the full range of temperature regimes, a standard protocol can be used to map any storage area in the facility.
3.2. The mapping protocol should contain the following sections:
- Approval page and change control history.
- Acronyms and glossary.
- Description and rational
- Scope
- Objectives.
- Methodology
- Mapping report template.
- Annexes as needed, including templates for the mapping report
3.3. Methodology:
- STEP 1 – select EDLMs:
- STEP 2 – designate the mapping team:
- STEP 3 – survey the site:
- STEP 4 – establish acceptance criteria:
- STEP 5 – determine EDLM locations:
- STEP 6 – record EDLM, monitoring sensors and thermostat locations:
- STEP 7 – label and program the EDLMs:
- STEP 8 – fix EDLMs in position:
- STEP 9 – conduct the mapping exercise:
- STEP 10 – download and consolidate the data:
3.4. Mapping report template:
The mapping report should include the following sections:
- Introduction: a description of the objectives of the mapping study.
- Summary: a summary and discussion of the results organised in the sequence set out in the mapping protocol, including a summary of deviations (if any).
- Conclusions and recommendations: a general conclusion for all verification’s and observations indicating the acceptability of the equipment for operating Recommendations and remarks can be incorporated as well.
- Report annexes: The report annexes should contain the following:
- The site survey, showing EDLM locations.
- The raw data, presented using the appropriate test data sheet format
- Spreadsheet data and related temperature graphs for every EDLM used in the mapping exercise.
- Raw results of the data analysis, including hot and cold spots.
- Key documents and notes prepared during the mapping exercise, together with any other supporting material.
- Deviation reports, including Corrective and Preventive Actions (CAPA) forms, if required. This may include a recommendation for partial or total re-mapping.Calibration certificates for all EDLMs used.
4.0. Implementing the mapping report recommendations:
The final outcome and purpose of a mapping exercise is the implementation of the report recommendations.
References
- Health Canada (HPFB Inspectorate): Guide 0069: Guidelines for temperature Control of Drug Products during Storage and Transportation. October 17, 2005. http://www.rxcritical.ca/pdf/Guide-0069.pdf
- IATA. 2013/2014 Perishable Cargo Regulations (ePCR) & Temperature Control
- Regulations (eTCR)
- http://www.iata.org/publications/Pages/temperature-control-regulations.aspx
- PDA Technical Report No. 39: Guidance for Temperature Controlled Medicinal Products: Maintaining the Quality of Temperature-Sensitive Medicinal Products through the Transportation Environment. Parenteral Drug Association. 2007. https://store.pda.org/ProductCatalog/Product.aspx?ID=1270
- United States Pharmaceopaedia: Chapter 1079: Good Storage & Shipping Practices. https://mc.usp.org/sites/default/files/documents/GeneralChapterPDFs/c1079%20USP36.pdf
- United States Pharmaceopaedia: Chapter 1118: Monitoring Devices – Time, Temperature and Humidity. http://www.pharmacopeia.cn/v29240/usp29nf24s0_c1118.html
- US Food and Drug Administration. Title 21–Food and Drugs. Chapter I–Food and Drug administration Department of Health and Human Services. Subchapter A— General. Part 11 Electronic Records; Electronic Signatures 21 CFR Part 11 http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?cfrpart=11.
- WHO Technical Report Series No. 961, 2011, Annex 9: Model guidance for the storage and transport of time- and temperature-sensitive pharmaceutical http://apps.who.int/medicinedocs/documents/s18683en/s18683en.pdf
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=28&origin=fwqrc.wordpress.com&obj_id=151501655-28-653e423aaea921 CommentGMP
CEP/COS
CEP/COS
Hi everyone!
Today we are going to discuss about the CEP/COS (Certification of suitability of European Pharmacopoeia monographs/ Certificate of suitability)!
Before filing a CEP, Let’s have a look about its history.
Image source: http://sociusgroup.net/2017/12/05/community-experience-project-cep-2/
European Directorate for the Quality of medicines and Healthcare was established in the year 1992 as a pilot scale and took over charge as a routine procedure in the year 1994 for the chemical substances. It was then expanded in the year 2003 and included herbal drugs and herbal drug preparations.
The aim of the EDQM is to assess whether the relevant European Pharmacopoeia monograph(s) can be used to adequately to control the quality and impurity profile of an API or excipient produced and/or distributed by the manufacturer. The procedure is complemented by an inspection programme of manufacturing and/or distribution sites, involving a network of around 100 assessors and 30 inspectors from 24 different national competent authorities and the EDQM.
Why to file a CEP?
The Certification procedure is not compulsory – it is a service that is offered to the API and excipient manufacturers, who can use a Certificate of Suitability (CEP) in an application for a new market authorisation (MAA) or for a variation of an existing MAA.

Image source: http://lagirlsweetea.blogspot.com/2012/02/
The manufacturer of a substance will be able to provide proof that the quality of the substance is suitably controlled by the relevant monographs of the European Pharmacopoeia by means of a certificate of suitability granted by the Certification Secretariat of the European Directorate for the Quality of Medicines (EDQM) To apply for a certificate a manufacturer will submit a detailed dossier (Refer ICH M4 CTD) which may also contain confidential data.
The procedure is intended to be applied for the assessment of quality with regards to the criteria of the monograph(s) as appropriate.
The certificate of suitability certifies that by applying the relevant monographs of the European Pharmacopoeia, if necessary with an annex appended to the certificate, it is possible to check whether or not the quality of the substance is suitable for use in medicinal products. In other words, it ensures that all possible impurities and contamination from this particular route of manufacture (including source materials), genotoxic impurities, elemental impurities that can be fully controlled.
The CEP certification procedure can be applied for the following substances:
1. Organic or inorganic substances (active or excipients), manufactured or extracted.
2. Substances produced by fermentation as indirect gene products, which are metabolites of microorganisms, irrespective of whether or not the microorganisms have been modified by traditional procedures or r-DNA technology (see the monograph Products of Fermentation).
3. Products with risk of transmitting agents of animal spongiform encephalopathies(TSE)
The certificate of suitability will be delivered in preference to the manufacturer of substances intended for pharmaceutical use. In special cases where the holder will not be the manufacturer but an authorised agent, a formal agreement is required.
The Certification procedure centralises the evaluation of data for the benefit of regulatory authorities and industry alike, thus saving time and resources. For example, on average it takes three days for a regulatory authority to assess an Active Substance Master File (ASMF), which is the document submitted by a manufacturer of a medicine as part of its application for MAA (the ASMF contains complete information on an API or finished drug dosage form). If the CEP included in the ASMF is used in 10 countries, this assessment only needs to be done once, thus saving 27 days and the corresponding resources.
Also, the Certification procedure provides the European Pharmacopoeia Commission with current information on the quality of substances on the European market, thus helping to identify whether or not a revision of specific European Pharmacopoeia monographs is needed.
CEPs – which are referred to in EU pharmaceutical legislation – are recognised by the European Pharmacopoeia member states and by a number of other countries and regions, such as Australia, Canada, New Zealand, Saudi Arabia, Singapore, South Africa, Taiwan and Tunisia. An increasing number of licensing authorities worldwide accept CEPs to support (fully or partially) the data related to the quality of APIs used in medicinal products.
In order for a specific manufacturer to be granted a CEP, the EDQM’s panel of assessors (drawn from national medicines agencies throughout Europe) review a detailed dossier submitted by the manufacturer.
This dossier describes the manufacturing process and the tests performed on the raw materials and on the substance produced, as well as the necessary in-process controls.
The manufacturer must demonstrate that its product complies with the quality standards required by the European Pharmacopoeia and EU legislation and, in particular, that the monograph can be used to control impurities. The applicant must also agree to comply with the relevant Good Manufacturing Practice (GMP) as defined in Part II of the EU GMP Guide, and to accept a site inspection at any time at the request of the application.
After complete assessment of the CEP, the manufacturer finally receives the CEP certificate, which is valid for 5 years. If there is a change in the manufacturing process or change in the content of the dossier, the CEP can be revised.
References:
• https://en.wikipedia.org/wiki/European_Directorate_for_the_Quality_of_Medicines
• COUNCIL OF EUROPE ,PUBLIC HEALTH COMMITTEE, RESOLUTION AP-CSP (07) 1
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=22&origin=fwqrc.wordpress.com&obj_id=151501655-22-653e4242a71361 Comment

Right Team For The Right Job at The Right Time
All the posts related to Life Sciences and its requirements shall be updated by the technical team on daily basis.
Everyone is open to review and share your comments regarding the posts.
Share this:
https://widgets.wp.com/likes/index.html?ver=20230906#blog_id=151501655&post_id=16&origin=fwqrc.wordpress.com&obj_id=151501655-16-653e4242aaea7Tagged Auditing, Dossier preparation, eCTD, EDQM, FDA, GMP, GMP Compliance, ISO standards, Quality Management System, Regulatory AffairsLeave a comment

For Quality & Regulatory Compliance
Thanks for joining me!
Good company in a journey makes the way seem shorter. — Izaak Walton

https://kwors.com
Dear AntoniDex
Thank you for your review comments.
We hope these tips help! Let us know if you have any other questions.